Alice Allen

Alice Allen
Member of Robinson College
PhD student in Prof Payne's group
Office: 524 Mott Bld
Phone: +44(0)1223 3 37459
Email: aa840 @ cam.ac.uk
TCM Group, Cavendish Laboratory
19 JJ Thomson Avenue,
Cambridge, CB3 0HE UK.
Research
Density functional theory (DFT) allows us to carry out electronic structure calculations for large biological systems, but cannot tell us about biological processes which take place on time scales longer than a few picoseconds due to the computational expense. To retain some of the accuracy of DFT calculations, whilst being able to access longer timescales, we use the electron density from the DFT calculation to parameterise classical biomolecular force fields for use in molecular dynamics simulations.
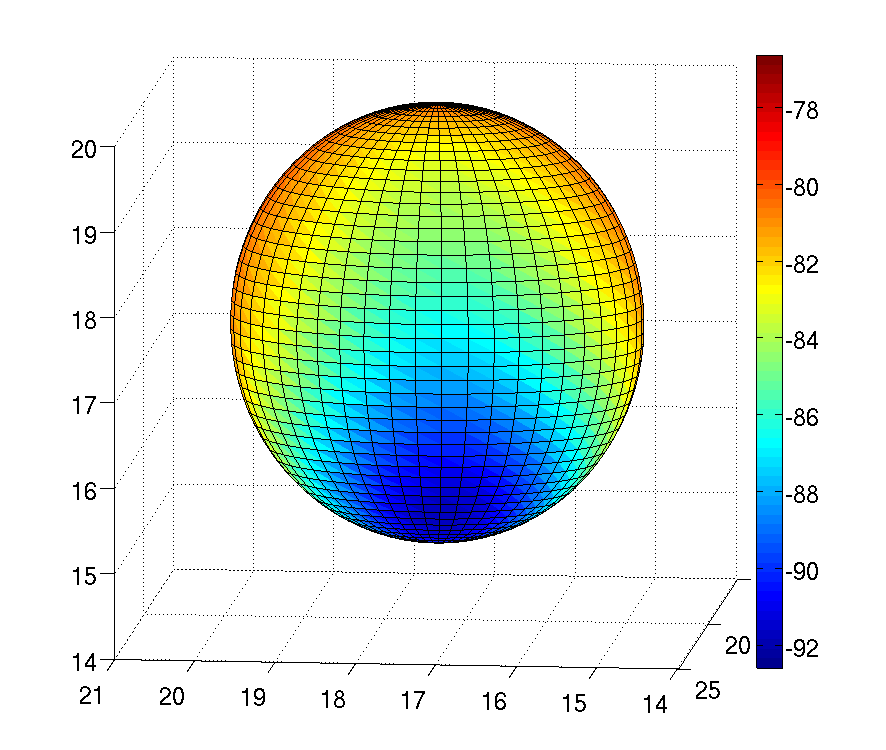
My work has focused on deriving the covalent bond and angles terms used, and reparameterising torsional parameters to be consistent with the new electrostatic and Lennard-Jones parameters. Additionally, I have looked at how off-center point charges can be used to recreate the quantum mechanical electrostatic potential for atoms with that anisotropic electron densities, due to characteristics such as σ holes and lone pairs. Anisotropy in the electrostatic potential of a nitrogen atom, due to a lone pair being present, is shown in the figure to the right.
In Plain English
Computational methods now exist to inform us about the structure and functions of biological molecules.Some techniques use quantum mechanics, however this only allows us to view the system for a very short amount of time (10-12 seconds) and biological processes can take place on a much longer time scale than this. Another method, classical molecular dynamics, can simulate systems on timescales a million times longer than quantum mechanics but lacks the same level of accuracy. The aim of this project is to use the information about forces in/between molecules from the quantum mechanical methods in classical molecular dynamics simulations. This will mean that we can simulate biological processes with increased accuracy in applications such as drug discovery.