Mike Payne

Prof Mike Payne FRS
Fellow of Pembroke College
Office: C3.013
Phone: +44(0)1223 3 37381
Email: mcp1 @ cam.ac.uk
Personal web site
TCM Group, Cavendish Laboratory
Ray Dolby Centre,
JJ Thomson Avenue,
Cambridge, CB3 0US UK.
Research Group
Research
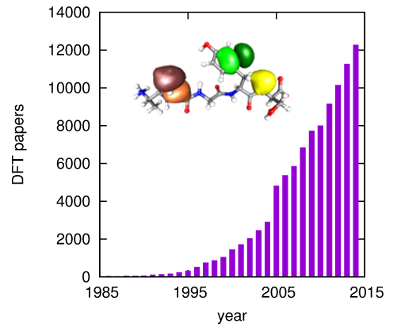
I have worked in the field of first principles quantum mechanical total energy calculations since 1985. I am the author of the total energy pseudopotential code, CASTEP (Cambridge Sequential Total Energy Package) the first such code that was accessible to non-experts and the first code to be commercialised. I am responsible for many of the technical developments of the methodology that led to the widespread adoption of density functional theory calculations based on the total energy pseudopotential technique. The remarkable impact of density functional theory is beautifully illustrated by the statistics in the figure on the right, which shows the number of publications per year based on this methodology.
More recently, I have been involved in the development of a range of other quantum mechanical methods, all of which are designed to retain the predictive capability and ease of use that has underpinned the success of CASTEP and which, I believe, will be critical ingredients in a predictive, black box multiscale modelling scheme. These methods are:
- ONETEP a linear scaling density functional theory code that allows calculations to be performed for many thousands of atoms on modest supercomputers.
- the Learn on the Fly hybrid (or QM/MM - quantum mechanics /molecular mechanics) modelling scheme developed in collaboration with Dr. Gábor Csányi in the Engineering Department in Cambridge and Professor Sandro De Vita in King’s College, London, and others. In this scheme, the regions of the system treated quantum mechanically can change with time and the choice of these regions can be delegated to the computer;
- the Gaussian Approximation Potential developed in collaboration with Dr. Gábor Csányi and Dr. Albert Bartok Partay in the Engineering Department in Cambridge. This is a new class of inter-atomic potential which does not assume a particular functional form but generates forces and energies using Gaussian approximation based on the forces and energies previously determined for a set of representative configurations. These energies and forces can be obtained from any source but usually will have been computed using first principles calculations.
I am also the Director of the EPSRC Centre for Doctoral Training in Computational Methods for Materials Science and the Chairman of the Cambridge High Performance Computing Service which has become a pioneer and innovator in many aspects of high performance computing and data.
Awards and Prizes:
1996 Maxwell Medal and Prize awarded by the Institute of Physics.
1998 Mott Lecture awarded by the Institute of Physics.
1999 Citation Superstar of the U.K., 1990-99 awarded by the Institute
for Scientific Information (23rd most
Highly cited Physical Scientist in the UK between 1990 and 1999 –
2,869 citations).
2008 Elected Fellow of the Royal Society.
2011 Elected Honorary Fellow of the Institute of Physics.
2014 Swan Medal awarded by the Institute of Physics.
Commercial Activities:
Consultant to Accelrys (1994-).
CASTEP (Cambridge Sequential Total
Energy Package) the set of programs I have developed
to perform total energy pseudopotential calculations is marketed by
Accelrys - cumulative sales
exceed $30 million and annual sales have exceeded £1million every year
since 1998.
Member of Technical Advisory Board of R.J. Mears, Inc (2003-6)
ONETEP a linear scaling density
functional theory code developed by the ODG (ONETEP
Developers Group) was licenced to Accelrys in 2004 – cumulative sales
now exceed £2million.
Featured Publications
- Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104 136403 (2010)
- Low-speed fracture instabilities in a brittle crystal Nature 455 1224 - U41 (2008)
- Introducing ONETEP: linear-scaling density functional simulations on parallel computers. J. Chem. Phys. 122 84119 (2005)
- First principles methods using CASTEP Z. Kristallogr. 220 567 - 570 (2005)
- Iterative Minimization Techniques For Abinitio Total-Energy Calculations - Molecular-Dynamics And Conjugate Gradients Rev. Mod. Phys. 64 1045 - 1097 (1992)