Can Koçer

Can Koçer
Member of King's College
PhD student in Dr Morris's group
Office: 525 Mott Bld
Phone: +44(0)1223 3 37358
Email: cpk27 @ cam.ac.uk
TCM Group, Cavendish Laboratory
19 JJ Thomson Avenue,
Cambridge, CB3 0HE UK.
Research
I develop first-principles computational methods and apply them to problems in materials science and condensed matter physics. My two main areas of focus are (1) density-functional theory (DFT) simulations of lithium-ion battery electrode materials, and (2) method development of phonon and electron-phonon coupling calculations for correlated materials with DFT+DMFT. Some highlights from my published work:
Complex Oxide Electrode Materials: A major focus of my research have been the Wadsley-Roth (crystallographic shear) phases, a promising class of Nb-based lithium-ion battery electrodes. These complex oxides have attracted attention for their ability to intercalate and deintercalate lithium very quickly, enabling fast-charging batteries. I have used DFT to study the electronic structure, cation disorder, lithium insertion mechanism, and lithium diffusion of these materials. These simulations led to detailed atomic-scale insights into the origin of the fast-charging behaviour by identifying the diffusion mechanism: a network of coupled, one-dimensional tunnels with low activation barriers allow rapid lithium motion.
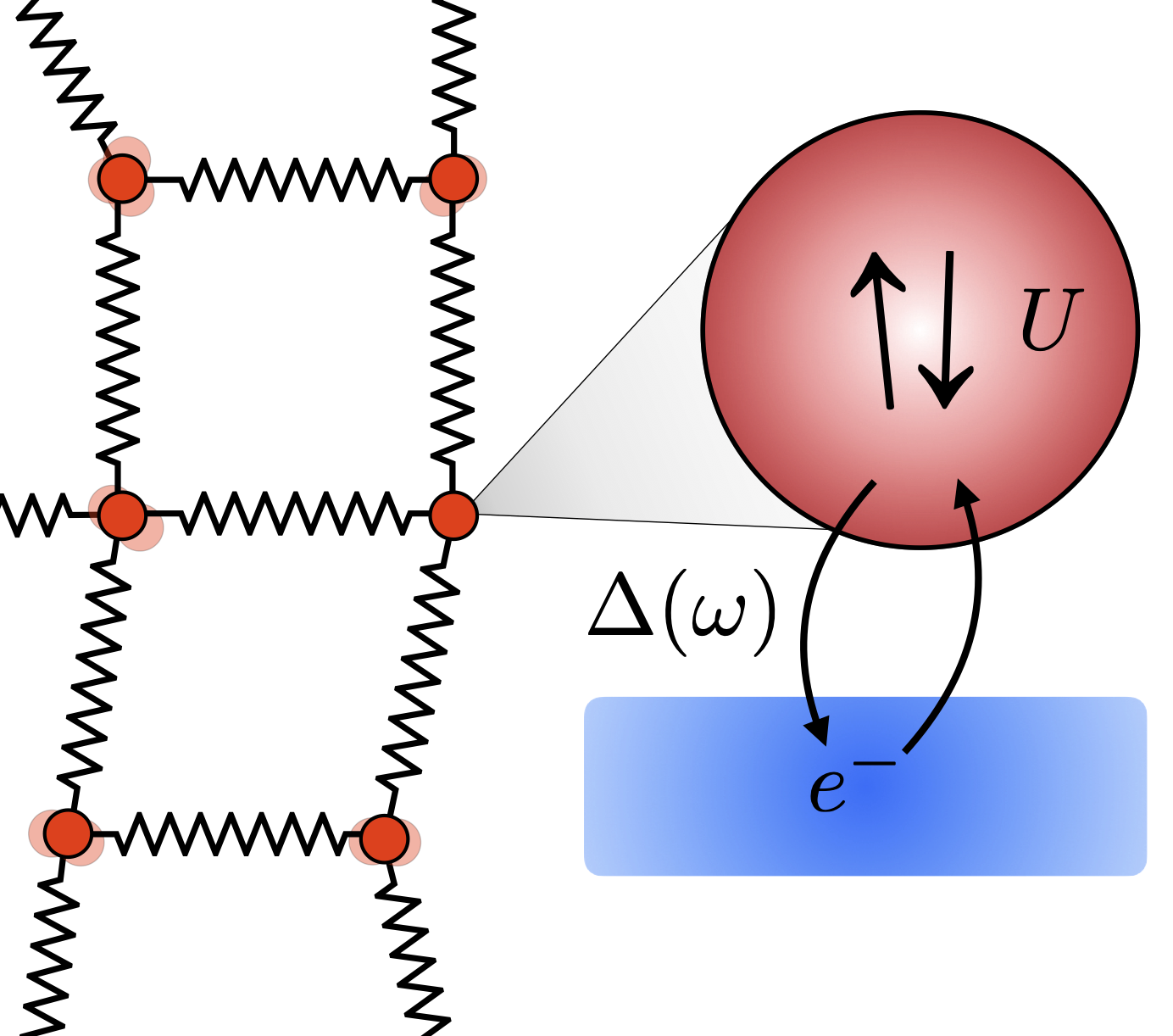
Lattice Dynamics with DFT+DMFT: Phonons play a fundamental role in the physics of solids. DFT calculations of phonons allow us to understand phonon-driven phenomena from first-principles, but their usefulness depends on how well the electronic structure of a material is described by DFT. We have developed a method to calculate phonons in correlated materials, using DFT+DMFT instead of DFT as the electronic structure method (see illustration on the right). This allows us to study temperature-driven effects on phonons that are purely due to electronic correlation, for example close to metal-insulator transitions.
In Plain English
I apply and develop computer simulation methods to study the properties of materials. Simulations of battery materials have been a major focus of my research. The insights we gained from these simulations help us understand experimental data and guide the design of future materials and devices. More recently, I have been working on developing new simulation methods for materials that are not well-described with current techniques.
Featured Publications
-
Efficient Lattice Dynamics Calculations for Correlated Materials with DFT+DMFT
Phys. Rev. B 102 245104 (2020) Editors' Suggestion -
Multimodal Structure Solution with 19F NMR Crystallography of Spin Singlet Molybdenum Oxyfluorides
J. Am. Chem. Soc. 142 12288 - 12298 (2020) -
Lithium Diffusion in Niobium Tungsten Oxide Shear Structures
Chem. Mater. 32 3980 - 3989 (2020) ACS Editors' Choice -
First-principles Study of Localized and Delocalized Electronic States in Crystallographic Shear Phases of Niobium Oxide
Phys. Rev. B 99 075151 (2019) -
Cation Disorder and Lithium Insertion Mechanism of Wadsley-Roth Crystallographic Shear Phases from First Principles
J. Am. Chem. Soc. 141 15121 - 15134 (2019) -
Ionic and Electronic Conduction in TiNb2O7
J. Am. Chem. Soc. 141 17606 - 17625 (2019)