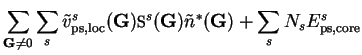


Next: 7.2 Energy gradients
Up: 7. Computational implementation
Previous: 7. Computational implementation
Contents
Subsections
A quantity which is frequently required is the overlap matrix
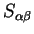 defined by
defined by
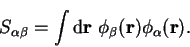 |
(7.3) |
The overlap matrix elements between the spherical-wave basis functions can
be calculated analytically (section 5.4),
and are denoted
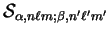 where
where
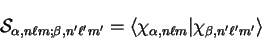 |
(7.4) |
so that the overlap matrix elements are given by
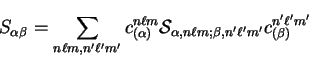 |
(7.5) |
recalling that the support functions may be assumed real in the case of
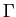 -point Brillouin zone sampling.
-point Brillouin zone sampling.
The kinetic energy of the non-interacting Kohn-Sham system is given by
![\begin{displaymath}
T_{\mathrm s}^{\mathrm J} = - \int {\mathrm d}{\bf r'}~
\lef...
...ight]_{{\bf r}={\bf r'}}
= 2 K^{\alpha \beta} T_{\beta \alpha}
\end{displaymath}](img930.gif) |
(7.6) |
in which
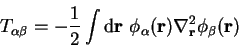 |
(7.7) |
are the matrix elements of the kinetic energy operator in the representation
of the support functions. Since all of the matrix elements between the
spherical-wave basis functions can be calculated analytically,
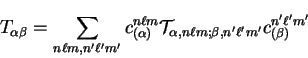 |
(7.8) |
where 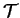 denotes matrix elements of the kinetic energy
operator between spherical-wave basis functions.
denotes matrix elements of the kinetic energy
operator between spherical-wave basis functions.
The Hartree and exchange-correlation terms are calculated by
determining the electronic density on a real-space
grid 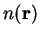 , and Fast Fourier Transforms (FFTs) are used to transform between
real- and reciprocal-space7.1to obtain
, and Fast Fourier Transforms (FFTs) are used to transform between
real- and reciprocal-space7.1to obtain
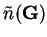 . The Hartree energy is then given by
. The Hartree energy is then given by
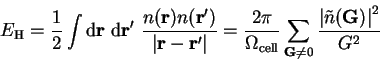 |
(7.9) |
where
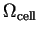 is the volume of the supercell and the
(infinite)
is the volume of the supercell and the
(infinite) 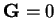 term is omitted because the system is charge neutral
overall. This term is therefore cancelled by similar terms in the ion-ion
and electron-ion interaction energies. The Hartree potential in real-space is
given by
term is omitted because the system is charge neutral
overall. This term is therefore cancelled by similar terms in the ion-ion
and electron-ion interaction energies. The Hartree potential in real-space is
given by
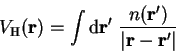 |
(7.10) |
but is calculated in reciprocal-space as
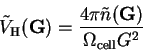 |
(7.11) |
and then transformed back into real-space by a FFT.
Having calculated the electron density on the grid points, the exchange-correlation energy is obtained by summing over those grid points
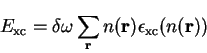 |
(7.12) |
in the local density approximation. 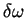 is the volume of the supercell divided by the number of grid points. The exchange-correlation potential is
similarly calculated at each grid point as
is the volume of the supercell divided by the number of grid points. The exchange-correlation potential is
similarly calculated at each grid point as
![\begin{displaymath}
V_{\mathrm {xc}}({\bf r}) = \left[ \frac{\mathrm d}{{\mathrm...
...\epsilon_{\mathrm {xc}}(n) \right\} \right]_{n = n({\bf r})} .
\end{displaymath}](img942.gif) |
(7.13) |
In practice, the values of
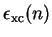 and
and
![$\frac{\mathrm d}{{\mathrm d}n} \left[
n \epsilon_{\mathrm {xc}}(n) \right]$](img944.gif) are tabulated for various values of
the electronic density
are tabulated for various values of
the electronic density 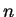 and then interpolated during the calculation.
and then interpolated during the calculation.
Like the Hartree potential, the local pseudopotential is also calculated in
reciprocal-space as
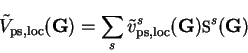 |
(7.14) |
where the summation is over ionic species 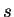 ,
,
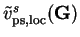 is the local pseudopotential for an isolated ion of species in
reciprocal-space and
is the local pseudopotential for an isolated ion of species in
reciprocal-space and
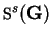 is the structure factor for
species defined by
is the structure factor for
species defined by
![\begin{displaymath}
{\mbox{\Bbb S}}^s({\bf G}) = \sum_{\alpha} \exp[-{\mathrm i}{\bf G} \cdot {\bf r}_{\alpha}^s]
\end{displaymath}](img949.gif) |
(7.15) |
where the sum is over all ions 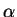 of species with positions
of species with positions
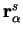 .
We note that in general the calculation of the structure factor is
an
.
We note that in general the calculation of the structure factor is
an 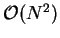 operation, but since it only has to be calculated once
for each atomic configuration, it is not a limiting factor of the overall
calculation at this stage. Within the quantum chemistry community,
work on generalised multipole expansions and new algorithms
[156,157,158,159,160,161,162]
has led to the development of methods to calculate Coulomb
interaction matrix elements which scale linearly with system-size.
The local pseudopotential energy can be calculated in reciprocal-space as
operation, but since it only has to be calculated once
for each atomic configuration, it is not a limiting factor of the overall
calculation at this stage. Within the quantum chemistry community,
work on generalised multipole expansions and new algorithms
[156,157,158,159,160,161,162]
has led to the development of methods to calculate Coulomb
interaction matrix elements which scale linearly with system-size.
The local pseudopotential energy can be calculated in reciprocal-space as
where
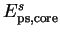 is the pseudopotential core energy, and
is the pseudopotential core energy, and
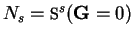 the number of ions of species . The matrix elements
the number of ions of species . The matrix elements
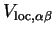 are defined by
are defined by
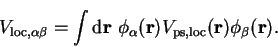 |
(7.17) |
The Hartree potential and local pseudopotential can be summed and then
transformed back
together into real-space and added to the exchange-correlation potential to
obtain the local part of the Kohn-Sham potential in real-space.
We note that the FFT is not strictly an 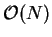 operation but an
operation but an
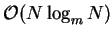 operation
(where
operation
(where 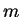 is some small number which depends upon the prime factors of the
number of grid points), but in practice (section 9.2)
this scaling is not observed.
is some small number which depends upon the prime factors of the
number of grid points), but in practice (section 9.2)
this scaling is not observed.
The non-local pseudopotential energy is given by
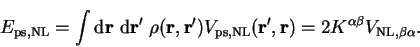 |
(7.18) |
The matrix elements of the non-local pseudopotential in the representation of
the support functions
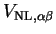 are calculated by
summing over all ions whose cores overlap the support regions of
are calculated by
summing over all ions whose cores overlap the support regions of
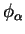 and
and 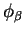 , and using the method described in
section 5.6.2 to calculate the spherical-wave basis function
matrix elements
, and using the method described in
section 5.6.2 to calculate the spherical-wave basis function
matrix elements
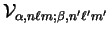 analytically. The result is therefore
analytically. The result is therefore
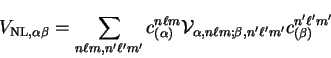 |
(7.19) |
which is of exactly the same form as the kinetic energy, so that in practice
the basis function matrix elements for the kinetic energy and non-local
pseudopotential are summed and the two contributions to the energy combined.
Next: 7.2 Energy gradients
Up: 7. Computational implementation
Previous: 7. Computational implementation
Contents
Peter Haynes