


Up: thesis
Previous: B. Conjugate gradients
Contents
- 1
-
J. S. Bell.
Speakable and unspeakable in quantum mechanics (Cambridge
University Press, 1987).
- 2
-
T. Kinoshita and W. B. Lindquist.
Eighth-order anomalous magnetic moment of the electron.
Phys. Rev. Lett. 47 (22), 1573 (November 1981).
- 3
-
Robert S. Van Dyck Jr., Paul B. Schwinberg and Hans G. Dehmelt.
New high-precision comparison of electron and positron 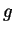 factors.
factors.
Phys. Rev. Lett. 59 (1), 26 (July 1987).
- 4
-
P. A. M. Dirac.
The Principles of Quantum Mechanics, chap. I, p. 15ff.
(Clarendon Press, Oxford, 1958), 4th edn.
- 5
-
Alastair I. M. Rae.
Quantum Mechanics, chap. 4, p. 63 (Adam Hilger, Bristol,
1986), 2nd edn.
- 6
-
M. Born and R. Oppenheimer.
Zur Quantentheorie der Molekeln.
Ann. Phys. (Leipzig) 84 (20), 457 (1927).
- 7
-
J. M. Ziman.
Principles of the Theory of Solids, chap. 6, pp. 200-203
(Cambridge University Press, 1972), 2nd edn.
- 8
-
Stephen Gasiorowicz.
Quantum Physics, chap. 16, p. 255ff. (John Wiley & Sons, New
York, 1974).
- 9
-
M. P. Allen and D. J. Tildesley.
Computer Simulation of Liquids, chap. 10, p. 270ff. (Oxford
University Press, 1987).
- 10
-
M. J. Gillan.
The quantum simulation of hydrogen in metals.
Phil. Mag. A 58 (1), 257 (1988).
- 11
-
R. Car and M. Parrinello.
Unified approach for molecular dynamics and density-functional
theory.
Phys. Rev. Lett. 55 (22), 2471 (November 1985).
- 12
-
T. A. Arias, M. C. Payne and J. D. Joannopoulos.
Ab initio molecular-dynamics techniques extended to
large-length-scale systems.
Phys. Rev. B 45 (4), 1538 (January 1992).
- 13
-
M. V. Berry and J. M. Robbins.
Indistinguishability for quantum particles: spin, statistics and the
geometric phase.
Proc. R. Soc. Lond. A 453, 1771 (1997).
- 14
-
L. D. Landau and E. M. Lifshitz.
Quantum Mechanics (Non-relativistic Theory), chap. IX, p.
241ff. (Pergamon Press, Oxford, 1973), 3rd edn.
- 15
-
V. B. Berestetskii, E. M. Lifshitz and L. P. Pitaevskii.
Quantum Electrodynamics, chap. II, III, pp. 33ff., 62ff.
(Pergamon Press, Oxford, 1979), 2nd edn.
- 16
-
P. Hohenberg and W. Kohn.
Inhomogeneous electron gas.
Phys. Rev. 136 (3B), 864 (November 1964).
- 17
-
Mel Levy.
Electron densities in search of Hamiltonians.
Phys. Rev. A 26 (3), 1200 (September 1982).
- 18
-
H. Englisch and R. Englisch.
Exact density functionals for ground-state energies.
Phys. Stat. Sol. (b) 123, 711 (1984).
- 19
-
H. Englisch and R. Englisch.
Exact density functionals for ground-state energies.
Phys. Stat. Sol. (b) 124, 373 (1984).
- 20
-
M. Levy.
Universal variational functionals of electron densities, first-order
density matrices, and natural spin-orbitals and solution of the
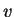 -representability problem.
-representability problem.
Proc. Natl. Acad. Sci. 76, 6062 (1979).
- 21
-
Mel Levy and John P. Perdew.
The constrained search formulation of density functional theory.
In Density Functional Methods in Physics (eds. Reiner M.
Dreizler and Joao da Providencia), p. 11ff. (Plenum Publishing Corporation,
New York, 1985).
- 22
-
T. L. Gilbert.
Hohenberg-Kohn theorem for nonlocal external potentials.
Phys. Rev. B 12 (6), 2111 (September 1975).
- 23
-
L. H. Thomas.
The calculation of atomic fields.
Proc. Camb. Phil. Soc. 23, 542 (November 1927).
- 24
-
E. Fermi.
Un metodo statistico per la determinazione di alcune proprietà
dell'atome.
Rend. Accad. Naz. Lincei 6, 602 (1927).
- 25
-
E. Fermi.
Eine statistische Methode zur Bestimmung einiger Eigenschaften
des Atoms und ihre Anwendung auf die Theorie des periodischen Systems
der Elemente.
Z. Phys. 48, 73 (1928).
- 26
-
E. Teller.
On the stability of molecules in the Thomas-Fermi theory.
Rev. Mod. Phys. 34, 627 (1962).
- 27
-
Elliott H. Lieb.
Thomas-Fermi and related theories of atoms and molecules.
Rev. Mod. Phys. 53 (4), 603 (October 1981).
- 28
-
Lin-Wang Wang and Michael P. Teter.
Kinetic-energy functional of the electron density.
Phys. Rev. B 45 (23), 13196 (June 1992).
- 29
-
M. Pearson, E. Smargiassi and P. A. Madden.
Ab initio molecular dynamics with an orbital-free density
functional.
J. Phys.: Condens. Matter 5, 3221 (1993).
- 30
-
F. Perrot.
Hydrogen-hydrogen interaction in an electron gas.
J. Phys.: Condens. Matter 6, 431 (1994).
- 31
-
Enrico Smargiassi and Paul A. Madden.
Orbital-free kinetic-energy functionals for first-principles
molecular dynamics.
Phys. Rev. B 49 (8), 5220 (February 1994).
- 32
-
Michael Foley and Paul A. Madden.
Further orbital-free kinetic-energy functionals for ab initio
molecular dynamics.
Phys. Rev. B 53 (16), 10589 (April 1996).
- 33
-
W. Kohn and L. J. Sham.
Self-consistent equations including exchange and correlation effects.
Phys. Rev. 140 (4A), 1133 (November 1965).
- 34
-
John P. Perdew and Mel Levy.
Extrema of the density functional for the energy: Excited states from
the ground-state theory.
Phys. Rev. B 31 (10), 6264 (May 1985).
- 35
-
D. M. Ceperley and B. J. Alder.
Ground state of the electron gas by a stochastic method.
Phys. Rev. Lett. 45 (7), 566 (August 1980).
- 36
-
J. P. Perdew and Alex Zunger.
Self-interaction correction to density-functional approximations for
many-electron systems.
Phys. Rev. B 23 (10), 5048 (May 1981).
- 37
-
R. O. Jones and O. Gunnarsson.
The density functional formalism, its applications and prospects.
Rev. Mod. Phys. 61 (3), 689 (July 1989).
- 38
-
J. Harris and R. O. Jones.
The surface energy of a bounded electron gas.
J. Phys. F 4, 1170 (August 1974).
- 39
-
D. C. Langreth and J. P. Perdew.
The exchange-correlation energy of a metallic surface.
Solid State Comm. 17, 1425 (1975).
- 40
-
J. Harris.
Adiabatic-connection approach to Kohn-Sham theory.
Phys. Rev. A 29 (4), 1648 (April 1984).
- 41
-
E. K. U. Gross, E. Runge and O. Heinonen.
Many-Particle Theory, chap. 16, p. 179ff. (Adam Hilger, New
York, 1991), English edn.
- 42
-
O. Gunnarsson and B. I. Lundqvist.
Exchange and correlation in atoms, molecules, and solids by the
spin-density-functional formalism.
Phys. Rev. B 13 (10), 4274 (May 1976).
- 43
-
David C. Langreth and M. J. Mehl.
Easily implementable nonlocal exchange-correlation energy functional.
Phys. Rev. Lett. 47 (6), 446 (August 1981).
- 44
-
David C. Langreth and M. J. Mehl.
Beyond the local-density approximation in calculations of
ground-state electronic properties.
Phys. Rev. B 28 (4), 1809 (August 1983).
- 45
-
Neil W. Ashcroft and N. David Mermin.
Solid State Physics, chap. 8, p. 132ff. (Saunders College,
Philadelphia, 1976), International edn.
- 46
-
J. L. Lebowitz and Elliott H. Lieb.
Existence of thermodynamics for real matter with Coulomb forces.
Phys. Rev. Lett. 22 (13), 631 (March 1969).
- 47
-
L. P. Bouckaert, R. Smoluchowski and E. Wigner.
Theory of Brillouin zones and symmetry properties of wave functions
in crystals.
Phys. Rev. 50, 58 (July 1936).
- 48
-
A. Baldereschi.
Mean-value point in the Brillouin zone.
Phys. Rev. B 7 (12), 5212 (June 1973).
- 49
-
D. J. Chadi and Marvin L. Cohen.
Special points in the Brillouin zone.
Phys. Rev. B 8 (12), 5747 (December 1973).
- 50
-
Hendrik J. Monkhorst and James D. Pack.
Special points for Brillouin-zone integrations.
Phys. Rev. B 13 (12), 5188 (June 1976).
- 51
-
D. J. Chadi.
Special points for Brillouin-zone integrations.
Phys. Rev. B 16 (4), 1746 (August 1977).
- 52
-
R. A. Evarestov and V. P. Smirnov.
Special points of the Brillouin zone and their use in the solid
state theory.
Phys. Stat. Sol. 119, 9 (1983).
- 53
-
Sverre Froyen.
Brillouin-zone integration by Fourier quadrature: Special points
for superlattice and supercell calculations.
Phys. Rev. B 39 (5), 3168 (February 1989).
- 54
-
I. J. Robertson and M. C. Payne.
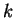 -point sampling and the
-point sampling and the 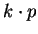 method in pseudopotential
total energy calculations.
method in pseudopotential
total energy calculations.
J. Phys.: Condens. Matter 2, 9837 (1990).
- 55
-
I. J. Robertson and M. C. Payne.
The method in pseudopotential total energy calculations:
error reduction and absolute energies.
J. Phys.: Condens. Matter 3, 8841 (1991).
- 56
-
James C. Phillips.
Energy-band interpolation scheme based on a pseudopotential.
Phys. Rev. 112 (3), 685 (November 1958).
- 57
-
James C. Phillips and Leonard Kleinman.
New method for calculating wave functions in crystals and molecules.
Phys. Rev. 116 (2), 287 (October 1959).
- 58
-
Volker Heine.
The Pseudopotential Concept, vol. 24 of Solid State
Physics, p. 1 (Academic Press, New York, 1970).
- 59
-
J. Ihm.
Total energy calculations in solid-state physics.
Rep. Prog. Phys. 51 (1), 105 (1988).
- 60
-
W. E. Pickett.
Pseudopotential methods in condensed matter applications.
Comp. Phys. Rep. 9 (3), 115 (1989).
- 61
-
Conyers Herring.
A new method for calculating wave functions in crystals.
Phys. Rev. 57, 1169 (June 1940).
- 62
-
Leonard I. Schiff.
Quantum Mechanics, chap. 5, p. 116ff. (McGraw-Hill,
Singapore, 1968), 3rd edn.
- 63
-
Michael Teter.
Additional condition for transferability in pseudopotentials.
Phys. Rev. B 48 (8), 5031 (August 1993).
- 64
-
A. Filippetti, David Vanderbilt, W. Zhong, Yong Cai and G. B. Bachelet.
Chemical hardness, linear response, and pseudopotential
transferability.
Phys. Rev. B 52 (16), 11793 (October 1995).
- 65
-
Antonio Redondo, William A. Goddard III and T. C. McGill.
Ab initio effective potentials for silicon.
Phys. Rev. B 15 (10), 5038 (May 1977).
- 66
-
D. R. Hamann, M. Schlüter and C. Chiang.
Norm-conserving pseudopotentials.
Phys. Rev. Lett. 43 (20), 1494 (November 1979).
- 67
-
Alex Zunger and Marvin L. Cohen.
First-principles nonlocal-pseudopotential approach in the
density-functional formalism. II. Application to electronic and
structural properties of solids.
Phys. Rev. B 20 (10), 4082 (November 1979).
- 68
-
G. P. Kerker.
Non-singular atomic pseudopotentials for solid state applications.
J. Phys. C 13 (9), L189 (March 1980).
- 69
-
G. B. Bachelet, D. R. Hamann and M. Schlüter.
Pseudopotentials that work: From H to Pu.
Phys. Rev. B 26 (8), 4199 (October 1982).
- 70
-
D. R. Hamann.
Generalized norm-conserving pseudopotentials.
Phys. Rev. B 40 (5), 2980 (August 1989).
- 71
-
Andrew M. Rappe, Karin M. Rabe, Efthimios Kaxiras and J. D. Joannopoulos.
Optimized pseudopotentials.
Phys. Rev. B 41 (2), 1227 (January 1990).
- 72
-
J. S. Lin, A. Qteish, M. C. Payne and V. Heine.
Optimized and transferable nonlocal separable ab initio
pseudopotentials.
Phys. Rev. B 47 (8), 4174 (February 1993).
- 73
-
Ming-Hsien Lee.
Advanced Pseudopotentials for Large Scale Electronic Structure
Calculations.
Ph.D. thesis, University of Cambridge, Cavendish Laboratory (1994).
- 74
-
N. Troullier and José Luís Martins.
Efficient pseudopotentials for plane-wave calculations.
Phys. Rev. B 43 (3), 1993 (January 1991).
- 75
-
David Vanderbilt.
Soft self-consistent pseudopotentials in a generalized eigenvalue
formalism.
Phys. Rev. B 41 (11), 7892 (April 1990).
- 76
-
Eric L. Shirley, Douglas C. Allan, Richard M. Martin and J. D. Joannopoulos.
Extended norm-conserving pseudopotentials.
Phys. Rev. B 40 (6), 3652 (August 1989).
- 77
-
Leonard Kleinman and D. M. Bylander.
Efficacious form for model pseudopotentials.
Phys. Rev. Lett. 48 (20), 1425 (May 1982).
- 78
-
Peter E. Blöchl.
Generalized separable potentials for electronic-structure
calculations.
Phys. Rev. B 41 (8), 5414 (March 1990).
- 79
-
J. Ihm, Alex Zunger and Marvin L. Cohen.
Momentum-space formalism for the total energy of solids.
J. Phys. C 12, 4409 (1979).
- 80
-
P. J. H. Denteneer and W. van Haeringen.
The pseudopotential-density-functional method in momentum space:
details and test cases.
J. Phys. C 18, 4127 (1985).
- 81
-
Michael P. Teter, Michael C. Payne and Douglas C. Allan.
Solution of Schrödinger's equation for large systems.
Phys. Rev. B 40 (18), 12255 (December 1989).
- 82
-
M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias and J. D. Joannopoulos.
Iterative minimization techniques for ab initio total-energy
calculations: molecular dynamics and conjugate gradients.
Rev. Mod. Phys. 64 (4), 1045 (October 1992).
- 83
-
G. Kresse and J. Furthmuller.
Efficient iterative schemes for ab-initio total-energy
calculations using a plane-wave basis-set.
Phys. Rev. B 54 (16), 11169 (1996).
- 84
-
X.-P. Li, R. W. Nunes and David Vanderbilt.
Density-matrix electronic-structure method with linear system-size
scaling.
Phys. Rev. B 47 (16), 10891 (April 1993).
- 85
-
S.-Y. Qiu, C. Z. Wang, K. M. Ho and C. T. Chan.
Tight-binding molecular dynamics with linear system-size scaling.
J. Phys.: Condens. Matter 6, 9153 (1994).
- 86
-
A. Canning, G. Galli, F. Mauri, A. de Vita and R. Car.
O(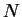 ) tight-binding molecular dynamics on massively parallel
computers: an orbital decomposition approach.
) tight-binding molecular dynamics on massively parallel
computers: an orbital decomposition approach.
Comp. Phys. Comm. 94, 89 (1996).
- 87
-
A. P. Horsfield, A. M. Bratkovsky, D. G. Pettifor and M. Aoki.
Bond-order potential and cluster recursion for the description of
chemical-bonds - efficient real-space methods for tight-binding
molecular-dynamics.
Phys. Rev. B 53 (3), 1656 (1996).
- 88
-
D. R. Bowler, M. Aoki, C. M. Goringe, A. P. Horsfield and D. G. Pettifor.
A comparison of linear scaling tight-binding methods.
Modelling Simul. Mater. Sci. Eng. 5 (3), 199 (1997).
- 89
-
Weitao Yang.
Direct calculation of electron density in density-functional theory.
Phys. Rev. Lett. 66 (11), 1438 (March 1991).
- 90
-
Weitao Yang.
A local projection method for the linear combination of atomic
orbital implementation of density-functional theory.
J. Chem. Phys. 94 (2), 1208 (January 1991).
- 91
-
Qingsheng Zhao and Weitao Yang.
Analytical energy gradients and geometry optimization in the
divide-and-conquer method for large molecules.
J. Chem. Phys. 102 (24), 9598 (June 1995).
- 92
-
Weitao Yang and Tai-Sung Lee.
A density-matrix divide-and-conquer approach for electronic structure
calculations of large molecules.
J. Chem. Phys. 103 (13), 5674 (October 1995).
- 93
-
Jian Ping Lu and Weitao Yang.
The shape of large single- and multiple-shell fullerenes.
Phys. Rev. B 49 (16), 11421 (April 1994).
- 94
-
Darrin M. York, Tai-Sung Lee and Weitao Yang.
Quantum mechanical study of aqueous polarization effects on
biological macromolecules.
J. Am. Chem. Soc. 118, 10940 (1996).
- 95
-
R. Haydock, Volker Heine and M. J. Kelly.
Electronic structure based on the local atomic environment for
tight-binding bands.
J. Phys. C 5, 2845 (1972).
- 96
-
Roger Haydock.
The Recursive Solution of the Schrödinger Equation,
vol. 35 of Solid State Physics, p. 215 (Academic Press, New York,
1980).
- 97
-
S. Baroni and P. Giannozzi.
Towards very large-scale electronic-structure calculations.
Europhys. Lett. 17 (6), 547 (February 1992).
- 98
-
David A. Drabold and Otto F. Sankey.
Maximum entropy approach for linear scaling in the electronic
structure problem.
Phys. Rev. Lett. 70 (23), 3631 (June 1993).
- 99
-
Lin-Wang Wang.
Calculating the density of states and optical-absorption spectra of
large quantum systems by the plane-wave moments method.
Phys. Rev. B 49 (15), 10154 (April 1994).
- 100
-
Otto F. Sankey, David A. Drabold and Andrew Gibson.
Projected random vectors and the recursion method in the
electronic-structure problem.
Phys. Rev. B 50 (3), 1376 (July 1994).
- 101
-
R. N. Silver and H. Röder.
Calculation of the densities of states and spectral functions by
Chebyshev recursion and maximum entropy.
Phys. Rev. E 56 (4), 4822 (October 1997).
- 102
-
Yang Wang, G. M. Stocks, W. A. Shelton, D. M. C. Nicholson, Z. Szotek and W. M.
Temmerman.
Order-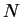 multiple scattering approach to electronic structure
calculations.
multiple scattering approach to electronic structure
calculations.
Phys. Rev. Lett. 75 (15), 2867 (October 1995).
- 103
-
I. A. Abrikosov, A. M. N. Niklasson, S. I. Simak, B. Johansson, A. V. Ruban and
H. L. Skriver.
Order- Green's function technique for local environment
effects in alloys.
Phys. Rev. Lett. 76 (22), 4203 (May 1996).
- 104
-
I. A. Abrikosov, S. I. Simak, B. Johansson, A. V. Ruban and H. L. Skriver.
Locally self-consistent Green's function approach to the electronic
structure problem.
Phys. Rev. B 56 (15), 9319 (October 1997).
- 105
-
S. Goedecker and L. Colombo.
Efficient linear scaling algorithm for tight-binding molecular
dynamics.
Phys. Rev. Lett. 73 (1), 122 (July 1994).
- 106
-
S. Goedecker and M. Teter.
Tight-binding electronic-structure calculations and tight-binding
molecular dynamics with localized orbitals.
Phys. Rev. B 51 (15), 9455 (April 1995).
- 107
-
Roi Baer and Martin Head-Gordon.
Chebyshev expansion methods for electronic structure calculations on
large molecular systems.
J. Chem. Phys. 107 (23), 10003 (December 1997).
- 108
-
Uwe Stephan and David A. Drabold.
Order- projection method for first-principles computations of
electronic quantities and Wannier functions.
Phys. Rev. B 57 (11), 6391 (March 1998).
- 109
-
S. Goedecker.
Integral representation of the Fermi distribution and its
applications in electronic-structure calculations.
Phys. Rev. B 48 (23), 17573 (December 1993).
- 110
-
D. M. C. Nicholson and X.-G. Zhang.
Approximate occupation functions for density-functional calculations.
Phys. Rev. B 56 (20), 12805 (November 1997).
- 111
-
Florian Gagel.
Finite-temperature evaluation of the Fermi density operator.
J. Comp. Phys. 139, 399 (1998).
- 112
-
A. F. Voter, J. D. Kress and R. N. Silver.
Linear-scaling tight binding from a truncated approach.
Phys. Rev. B 53 (19), 12733 (May 1996).
- 113
-
R. N. Silver, H. Roeder, A. F. Voter and J. D. Kress.
Kernel polynomial approximations for densities of states and spectral
functions.
J. Comp. Phys. 124, 115 (1996).
- 114
-
Giulia Galli and Michele Parrinello.
Large scale electronic structure calculations.
Phys. Rev. Lett. 69 (24), 3547 (December 1992).
- 115
-
Francesco Mauri, Giulia Galli and Roberto Car.
Orbital formulation for electronic-structure calculations with linear
system-size scaling.
Phys. Rev. B 47 (15), 9973 (April 1993).
- 116
-
Francesco Mauri and Giulia Galli.
Electronic-structure calculations and molecular-dynamics simulations
with linear system-size scaling.
Phys. Rev. B 50 (7), 4316 (August 1994).
- 117
-
Pablo Ordejón, David A. Drabold, Matthew P. Grumbach and Richard M. Martin.
Unconstrained minimization approach for electronic computations that
scales linearly with system size.
Phys. Rev. B 48 (19), 14646 (November 1993).
- 118
-
Pablo Ordejón, David A. Drabold, Richard M. Martin and Matthew P. Grumbach.
Linear system-size scaling methods for electronic-structure
calculations.
Phys. Rev. B 51 (3), 1456 (January 1995).
- 119
-
Jeongnim Kim, Francesco Mauri and Giulia Galli.
Total-energy global optimizations using nonorthogonal localized
orbitals.
Phys. Rev. B 52 (3), 1640 (July 1995).
- 120
-
K. C. Pandey, A. R. Williams and J. F. Janak.
Localized orbital theory of electronic structure: A simple
application.
Phys. Rev. B 52 (20), 14415 (November 1995).
- 121
-
Pablo Ordejón, Emilio Artacho and José M. Soler.
Self-consistent order- density-functional calculations for very
large systems.
Phys. Rev. B 53 (16), 10441 (April 1996).
- 122
-
Jeongnim Kim, John W. Wilkins, Furrukh S. Khan and Andrew Canning.
Extended Si {311} defects.
Phys. Rev. B 55 (24), 16186 (June 1997).
- 123
-
Giulia Galli.
Linear scaling methods for electronic structure calculations and
quantum molecular dynamics simulations.
Current Opinion in Solid State and Materials Science
1 (6), 864 (1996).
- 124
-
E. B. Stechel, A. R. Williams and Peter J. Feibelman.
-scaling algorithm for density-functional calculations of metals
and insulators.
Phys. Rev. B 49 (15), 10088 (April 1994).
- 125
-
W. Hierse and E. B. Stechel.
Order- methods in self-consistent density-functional
calculations.
Phys. Rev. B 50 (24), 17811 (December 1994).
- 126
-
E. Hernández and M. J. Gillan.
Self-consistent first-principles technique with linear scaling.
Phys. Rev. B 51 (15), 10157 (April 1995).
- 127
-
E. Hernández, M. J. Gillan and C. M. Goringe.
Linear-scaling density-functional-theory technique: The
density-matrix approach.
Phys. Rev. B 53 (11), 7147 (March 1996).
- 128
-
John M. Millam and Gustavo E. Scuseria.
Linear scaling conjugate gradient density matrix search as an
alternative to diagonalization for first principles electronic structure
calculations.
J. Chem. Phys. 106 (13), 5569 (April 1997).
- 129
-
Andrew D. Daniels, John M. Millam and Gustavo E. Scuseria.
Semiempirical methods with conjugate gradient density-matrix search
to replace diagonalization for molecular systems containing thousands of
atoms.
J. Chem. Phys. 107 (2), 425 (July 1997).
- 130
-
Karl Blum.
Density Matrix Theory and Applications, chap. 2, p. 37ff.
(Plenum Press, New York, 1981).
- 131
-
J. F. Janak.
Proof that
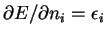 in
density-functional theory.
in
density-functional theory.
Phys. Rev. B 18 (12), 7165 (December 1978).
- 132
-
M. Weinert and J. W. Davenport.
Fractional occupations and density-functional energies and forces.
Phys. Rev. B 45 (23), 13709 (June 1992).
- 133
-
M. M. Valiev and G. W. Fernando.
Occupation numbers in density-functional calculations.
Phys. Rev. B 52 (15), 10697 (October 1995).
- 134
-
R. McWeeny.
Some recent advances in density matrix theory.
Rev. Mod. Phys. 32 (2), 335 (April 1960).
- 135
-
W. Kohn.
Density functional and density matrix method scaling linearly with
the number of atoms.
Phys. Rev. Lett. 76 (17), 3168 (April 1996).
- 136
-
W. Kohn.
Analytic properties of Bloch waves and Wannier functions.
Phys. Rev. 115 (4), 809 (August 1959).
- 137
-
E. I. Blount.
Formalisms of Band Theory, vol. 13 of Solid State
Physics, p. 305 (Academic Press, New York, 1962).
- 138
-
Jacques des Cloizeaux.
Energy bands and projection operators in a crystal: Analytic and
asymptotic properties.
Phys. Rev. 135 (3A), 685 (August 1964).
- 139
-
Jacques des Cloizeaux.
Analytical properties of 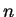 -dimensional energy bands and Wannier
functions.
-dimensional energy bands and Wannier
functions.
Phys. Rev. 135 (3A), 698 (August 1964).
- 140
-
Roi Baer and Martin Head-Gordon.
Sparsity of the density matrix in Kohn-Sham density functional
theory and an assessment of linear system-size scaling methods.
Phys. Rev. Lett. 79 (20), 3962 (November 1997).
- 141
-
P. E. Maslen, C. Ochsenfeld, C. A. White, M. S. Lee and M. Head-Gordon.
Locality and sparsity of ab initio one-particle density matrices and
localized orbitals.
J. Phys. Chem. 102, 2215 (1998).
- 142
-
Sohrab Ismail-Beigi and Tomás Arias.
On the locality of physics in metals, semiconductors, and insulators.
Phys. Rev. Lett. submitted.
- 143
-
Gregory H. Wannier.
The structure of electronic excitation levels in insulating crystals.
Phys. Rev. 52, 191 (August 1937).
- 144
-
S. F. Boys.
A general method of calculation for the stationary states of any
molecular system.
Proc. R. Soc. Lond. A 200, 542 (1950).
- 145
-
S. Obara and A. Saika.
Efficient recursive computation of molecular integrals over
Cartesian Gaussian functions.
J. Chem. Phys. 84 (7), 3963 (April 1986).
- 146
-
Otto F. Sankey and David J. Niklewski.
Ab initio multicenter tight-binding model for
molecular-dynamics simulations and other applications in covalent systems.
Phys. Rev. B 40 (6), 3979 (August 1989).
- 147
-
E. Hernández, M. J. Gillan and C. M. Goringe.
Basis functions for linear-scaling first-principles calculations.
Phys. Rev. B 55 (20), 13485 (May 1997).
- 148
-
Ross A. Lippert, T. A. Arias and Alan Edelman.
Multiscale computation with interpolating wavelets.
J. Comp. Phys. 140, 278 (1998).
- 149
-
James R. Chelikowsky, N. Troullier and Y. Saad.
Finite-difference-pseudopotential method: Electronic structure
calculations without a basis.
Phys. Rev. Lett. 72 (8), 1240 (February 1994).
- 150
-
P. D. Haynes and M. C. Payne.
Localised spherical-wave basis set for 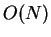 total-energy
pseudopotential calculations.
total-energy
pseudopotential calculations.
Comp. Phys. Comm. 102 (1-3), 17 (June 1997).
- 151
-
R. Courant and D. Hilbert.
Methods of Mathematical Physics, vol. 1, p. 535ff.
(Interscience Publishers, New York, 1953), 1st edn.
- 152
-
R. D. King-Smith, M. C. Payne and J. S. Lin.
Real-space implementation of nonlocal pseudopotentials for
first-principles total-energy calculations.
Phys. Rev. B 44 (23), 13063 (December 1991).
- 153
-
S. Goedecker.
Electronic structure methods exhibiting linear scaling of the
computational effort with respect to the size of the system.
Rev. Mod. Phys. submitted.
- 154
-
Y. Saad.
Iterative methods for sparse linear systems (PWS Publishing
Co., Boston, 1996).
- 155
-
D. R. Bowler and M. J. Gillan.
Length-scale ill conditioning in linear-scaling DFT.
Comp. Phys. Comm. 112 (2-3), 103 (1998).
- 156
-
Leslie Greengard.
Fast algorithms for classical physics.
Science 265, 909 (August 1994).
- 157
-
Christopher A. White, Benny G. Johnson, Peter M. W. Gill and Martin
Head-Gordon.
The continuous fast multipole method.
Chem. Phys. Lett. 230, 8 (November 1994).
- 158
-
Matthew C. Strain, Gustavo E. Scuseria and Michael J. Frisch.
Achieving linear scaling for the electronic quantum Coulomb
problem.
Science 271, 51 (January 1996).
- 159
-
Christopher A. White, Benny G. Johnson, Peter M. W. Gill and Martin
Head-Gordon.
Linear scaling density functional calculations via the continuous
fast multipole method.
Chem. Phys. Lett. 253, 268 (May 1996).
- 160
-
Ross D. Adamson, Jeremy P. Dombroski and Peter M. W. Gill.
Chemistry without Coulomb tails.
Chem. Phys. Lett. 254, 329 (May 1996).
- 161
-
José M. Pérez-Jordá and Weitao Yang.
Fast evaluation of the Coulomb energy for electron densities.
J. Chem. Phys. 107 (4), 1218 (July 1997).
- 162
-
S. Goedecker and O. V. Ivanov.
Linear scaling solution of the Coulomb problem using wavelets.
Solid State Comm. 105 (11), 665 (1998).
- 163
-
Christopher A. White, Paul Maslen, Michael S. Lee and Martin Head-Gordon.
The tensor properties of energy gradients within a non-orthogonal
basis.
Chem. Phys. Lett. 276, 133 (September 1997).
- 164
-
Nicola Marzari.
Ab initio Molecular Dynamics for Metallic Systems.
Ph.D. thesis, University of Cambridge, Cavendish Laboratory (1996).
- 165
-
Nicola Marzari, David Vanderbilt and M. C. Payne.
Ensemble density-functional theory for ab initio molecular
dynamics of metals and finite-temperature insulators.
Phys. Rev. Lett. 79 (7), 1337 (August 1997).
- 166
-
Daniel Sánchez-Portal, Emilio Artacho and José M. Soler.
Projection of plane-wave calculations into atomic orbitals.
Solid State Comm. 95 (10), 685 (1995).
- 167
-
Daniel Sánchez-Portal, Emilio Artacho and José M. Soler.
Analysis of atomic orbital basis sets from the projection of
plane-wave results.
J. Phys.: Condens. Matter 8, 3859 (1996).
- 168
-
M. D. Segall, C. J. Pickard, R. Shah and M. C. Payne.
Population analysis in plane wave electronic structure calculations.
Mol. Phys. 89 (2), 571 (1996).
- 169
-
W. Hierse and E. B. Stechel.
Robust localized-orbital transferability using the Harris
functional.
Phys. Rev. B 54 (23), 16515 (December 1996).
- 170
-
Pablo Fernández, Andrea Dal Corso, Alfonso Baldereschi and Francesco Mauri.
First-principles wannier functions of silicon and gallium arsenide.
Phys. Rev. B 55 (4), 1909 (January 1997).
- 171
-
Nicola Marzari and David Vanderbilt.
Maximally localized generalized Wannier functions for composite
energy bands.
Phys. Rev. B 56 (20), 12847 (November 1997).
- 172
-
Walter Kohn.
Density functional/Wannier function theory for systems of very many
atoms.
Chem. Phys. Lett. 208 (3,4), 167 (June 1993).
- 173
-
A. P. Sutton, M. W. Finnis, D. G. Pettifor and Y. Ohta.
The tight-binding bond model.
J. Phys. C 21, 35 (1988).
- 174
-
Jim Asher, Owen C. Jones, John G. Noyes and Geoffrey F. Phillips, eds.
Kaye & Laby's Tables of Physical and Chemical Constants,
pp. 45, 214 (Longman, Harlow, Essex, 1995), 16th edn.
- 175
-
P. P. Ewald.
Zur Begründung der Kristalloptik.
Ann. Phys. (Leipzig) 54 (23), 519 (1917).
- 176
-
P. P. Ewald.
Zur Begründung der Kristalloptik.
Ann. Phys. (Leipzig) 54 (24), 557 (1917).
- 177
-
P. P. Ewald.
Die Berechnung optischer und elektrostatischer Gitterpotentiale.
Ann. Phys. (Leipzig) 64, 253 (1921).
- 178
-
J. M. Ziman.
Principles of the Theory of Solids, chap. 2, pp. 37-42
(Cambridge University Press, Cambridge, 1972), 2nd edn.
- 179
-
Kai-Ming Ho, J. Ihm and J. D. Joannopoulos.
Dielectric matrix scheme for fast convergence in self-consistent
electronic-structure calculations.
Phys. Rev. B 25 (6), 4260 (March 1982).
- 180
-
P. H. Dederichs and R. Zeller.
Self-consistency iterations in electronic-structure calculations.
Phys. Rev. B 28 (10), 5462 (November 1983).
- 181
-
G. P. Kerker.
Efficient iteration scheme for self-consistent pseudopotential
calculations.
Phys. Rev. B 23 (6), 3082 (March 1981).
- 182
-
H. Hellmann.
Einfuhrung in die Quantumchemie (Deuticke, Leipzig, 1937).
- 183
-
R. P. Feynman.
Forces in molecules.
Phys. Rev. 56, 340 (August 1939).
- 184
-
P. Pulay.
Ab initio calculation of force constants and equilibrium
geometries in polyatomic molecules. i. theory.
Mol. Phys. 17 (2), 197 (1969).
- 185
-
C. M. Goringe, E. Hernández, M. J. Gillan and I. J. Bush.
Linear-scaling DFT-pseudopotential calculations on parallel
computers.
Comp. Phys. Comm. 102 (1-3), 1 (1997).
- 186
-
M. Abramowitz and I. Stegun.
Handbook of Mathematical Functions, chap. 10, p. 435 (Dover,
New York, 1965).
- 187
-
R. Fletcher and C. M. Reeves.
Function minimisation by conjugate gradients.
Comp. J. 7, 149 (1964).
- 188
-
D. M. Greig.
Optimisation, chap. 2, p. 41ff. (Longman, London, 1980).
- 189
-
E. Polak.
Computational Methods in Optimisation (Academic Press, 1971).
Peter Haynes