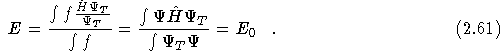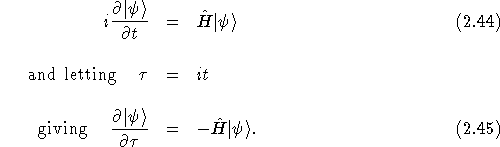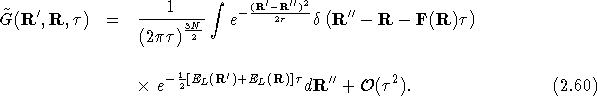The basis of DMC is to write the Schrödinger equation in imaginary time, taking
![]() can be expanded as:
can be expanded as:
where
![]()
The ![]() 's and
's and ![]() 's are the energy eigenvectors and
eigenvalues respectively of the time-independent Schrödinger equation.
's are the energy eigenvectors and
eigenvalues respectively of the time-independent Schrödinger equation.
We can write down the formal solution of the imaginary-time
Schrödinger equation, Eq.(![]() ),
),
noting that ![]() is the imaginary-time evolution
operator. Furthermore, expressing
is the imaginary-time evolution
operator. Furthermore, expressing ![]() in the form of
Eq.(
in the form of
Eq.(![]() ) implies that
) implies that
![]()
Letting ![]() be
be ![]() and hence
and hence ![]() , the evolution time for the system implies that, as long as
, the evolution time for the system implies that, as long as
![]() is not orthogonal to the ground-state,
is not orthogonal to the ground-state, ![]() , then
, then
where ![]() is the ground-state energy. This can be seen by
remembering that all other states have higher energies,
is the ground-state energy. This can be seen by
remembering that all other states have higher energies, ![]() ,
than the groundstate energy
,
than the groundstate energy ![]() , and will therefore decay
away faster. In the
, and will therefore decay
away faster. In the ![]() representation, Eq.(
representation, Eq.(![]() )
becomes:
)
becomes:
![]()
where
![]()
We now introduce an arbitrary energy offset term ![]() , such that the
imaginary-time Schrödinger equation, Eq.(
, such that the
imaginary-time Schrödinger equation, Eq.(![]() ), is recast as:
), is recast as:
Then if ![]() is adjusted to be the true ground-state energy,
is adjusted to be the true ground-state energy, ![]() ,
the asymptotic solution is a steady-state solution.
,
the asymptotic solution is a steady-state solution.
We now use the fact that the Schrödinger equation in imaginary time
looks like the diffusion equation. Explicitly writing out the
Hamiltonian in Eq.(![]() ), gives:
), gives:
This is just a 3N-dimensional diffusion equation, with ![]() playing the role of the density of diffusing
particles. The
playing the role of the density of diffusing
particles. The ![]() term is a
rate term, and describes the branching (or creation/annihilation)
processes. The entire equation can be simulated by a combination of a
diffusion and branching processes, in which the number of diffusing particles
increases or decreases at a given point proportional to the density of
diffusers and the potential energy at that point in configuration space.
term is a
rate term, and describes the branching (or creation/annihilation)
processes. The entire equation can be simulated by a combination of a
diffusion and branching processes, in which the number of diffusing particles
increases or decreases at a given point proportional to the density of
diffusers and the potential energy at that point in configuration space.
It turns out that solving Eq.(![]() ) this way is a very
inefficient way to simulate the Schrödinger equation on a computer.
This is because the branching rate, which is proportional to
) this way is a very
inefficient way to simulate the Schrödinger equation on a computer.
This is because the branching rate, which is proportional to ![]() , can diverge to
, can diverge to ![]() for systems of particles interacting via the Coulomb
interaction. This leads to large fluctuations in the number of
diffusing particles which leads to a large variance in the estimate of
the energy. These fluctuations can be dramatically reduced by the
introduction of importance sampling [28] in a similar way to the
implementation in the VMC algorithm (see section
for systems of particles interacting via the Coulomb
interaction. This leads to large fluctuations in the number of
diffusing particles which leads to a large variance in the estimate of
the energy. These fluctuations can be dramatically reduced by the
introduction of importance sampling [28] in a similar way to the
implementation in the VMC algorithm (see section ![]() ).
).
We will follow the scheme of Ref.[29] for the introduction of
importance sampling. The first step is to introduce a guiding
function, ![]() . We now define a new distribution
. We now define a new distribution ![]() which, if
which, if ![]() satisfies the Schrödinger equation, is also a solution
of the Schrödinger equation. Substituting
satisfies the Schrödinger equation, is also a solution
of the Schrödinger equation. Substituting ![]() into Eq.(
into Eq.(![]() ) yields
) yields
Where ![]() can now be interpreted as a ``quantum force''. We
follow [30] in defining this term as
can now be interpreted as a ``quantum force''. We
follow [30] in defining this term as
![]()
and ![]() , the branching term as
, the branching term as
![]()
which is defined in terms of the local energy of the guiding function
![]()
We now have a drift-diffusion equation for ![]() . The
branching term is proportional to the ``excess local energy'',
. The
branching term is proportional to the ``excess local energy'',
![]() , which with a good choice of
, which with a good choice of ![]() need not
become singular when
need not
become singular when ![]() does. To control branching we
need to choose
does. To control branching we
need to choose ![]() such that
such that ![]() is everywhere as smooth as
possible, i.e. we want as little variance as possible in
is everywhere as smooth as
possible, i.e. we want as little variance as possible in ![]() . Methods for optimising trial wavefunctions with exactly
this property are described in detail in chapter
. Methods for optimising trial wavefunctions with exactly
this property are described in detail in chapter ![]() .
In general, the trial wavefunction, Eq.(
.
In general, the trial wavefunction, Eq.(![]() ), used as
the input to a VMC calculation, makes a suitable choice of guiding
wavefunction.
), used as
the input to a VMC calculation, makes a suitable choice of guiding
wavefunction.
As well as reducing the fluctuations in the number of diffusing
particles, ![]() also has another important role for fermionic
systems. It determines the position of the nodes of the final
wavefunction, due to the necessity of using the fixed-node
approximation where the nodal structure of the exact groundstate
wavefunction is assumed to be the same as the nodal structure of
also has another important role for fermionic
systems. It determines the position of the nodes of the final
wavefunction, due to the necessity of using the fixed-node
approximation where the nodal structure of the exact groundstate
wavefunction is assumed to be the same as the nodal structure of
![]() to ensure that f is always of the same sign (see section
to ensure that f is always of the same sign (see section
![]() ). The accuracy of the position of the nodes in
). The accuracy of the position of the nodes in ![]() therefore determines how good the estimate of the ground-state energy,
therefore determines how good the estimate of the ground-state energy,
![]() , is. This can be seen by considering the fact that at long
(imaginary) times the distribution
, is. This can be seen by considering the fact that at long
(imaginary) times the distribution ![]() approaches
approaches
![]() , up to the constraint (imposed by the
fixed-node approximation) that
, up to the constraint (imposed by the
fixed-node approximation) that ![]() must vanish at the
nodes of
must vanish at the
nodes of ![]() . This implies that the long-time limit is
the true fermionic ground-state if and only if the nodes of
. This implies that the long-time limit is
the true fermionic ground-state if and only if the nodes of
![]() correspond to the exact nodes of the ground-state
wavefunction. The fixed-node energy is an upper bound to the
exact fermionic energy[31]. We simulate the equation
within only a small number of nodal regions. Each walker moves within
one nodal region and rejects all moves that attempt to cross the nodal
surface into another nodal region. This contradicts the requirement
that we stipulated earlier that the walk must be ergodic. The tiling
theorem
[32, 33], however, states that the nodal regions of
the true ground-state eigenfunction of a system of identical fermions
are all related by permutation symmetry. Furthermore the nodal regions
of the determinant of LDA eigenfunctions are also related by
permutation symmetry because the wavefunction is the ground-state of a
Hamiltonian, although it is not the many-body Hamiltonian of the
system we are interested in. This means that we can simulate the
equation within one nodal region and still be guaranteed to obtain the
``best'' variational result.
correspond to the exact nodes of the ground-state
wavefunction. The fixed-node energy is an upper bound to the
exact fermionic energy[31]. We simulate the equation
within only a small number of nodal regions. Each walker moves within
one nodal region and rejects all moves that attempt to cross the nodal
surface into another nodal region. This contradicts the requirement
that we stipulated earlier that the walk must be ergodic. The tiling
theorem
[32, 33], however, states that the nodal regions of
the true ground-state eigenfunction of a system of identical fermions
are all related by permutation symmetry. Furthermore the nodal regions
of the determinant of LDA eigenfunctions are also related by
permutation symmetry because the wavefunction is the ground-state of a
Hamiltonian, although it is not the many-body Hamiltonian of the
system we are interested in. This means that we can simulate the
equation within one nodal region and still be guaranteed to obtain the
``best'' variational result.
Eq.(![]() ) can be written in integral form, in doing this we
follow the procedure of Ref. [30]:
) can be written in integral form, in doing this we
follow the procedure of Ref. [30]:
where ![]() is the Green's function for the case
is the Green's function for the case ![]() . The
energy shift
. The
energy shift ![]() plays the role of an arbitrary
time-dependent renormalisation, chosen in such a way that the
probability distribution f remains finite and non-vanishing in the
limit
plays the role of an arbitrary
time-dependent renormalisation, chosen in such a way that the
probability distribution f remains finite and non-vanishing in the
limit ![]() .
.
The three terms on the left-hand side of Eq.(![]() ) describe
respectively, diffusion, drift and growth/decay. An approximate
Green's function can be formed, with an error of
) describe
respectively, diffusion, drift and growth/decay. An approximate
Green's function can be formed, with an error of ![]() for small
for small ![]() , by the product of Green's functions for diffusion,
drift and growth/decay[34]:
, by the product of Green's functions for diffusion,
drift and growth/decay[34]:
In order to deal with the nodes in the fermion ground-state we must
use the fixed-node approximation, as stated earlier (this and the
various schemes to try to improve upon it are discussed in section
![]() ). What this actually entails is that if a move is such
that a walker would cross the node then it is immediately rejected.
In other words, the node acts as an infinite potential barrier. It
should be noted that the use of importance sampling does not introduce
any extra approximations beyond the fixed node approximation already
mentioned. With the exception of the fixed node approximation it is
possible to calculate exact energies (and expectation values of any
other operator that commutes with the Hamiltonian,
). What this actually entails is that if a move is such
that a walker would cross the node then it is immediately rejected.
In other words, the node acts as an infinite potential barrier. It
should be noted that the use of importance sampling does not introduce
any extra approximations beyond the fixed node approximation already
mentioned. With the exception of the fixed node approximation it is
possible to calculate exact energies (and expectation values of any
other operator that commutes with the Hamiltonian, ![]() ) from the
distribution f, using
) from the
distribution f, using