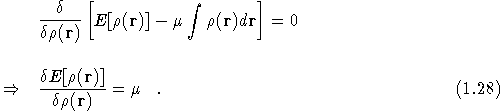Kohn and Sham [9] introduced a method based on the
Hohenberg-Kohn theorem that enables one to minimise the functional
![]() by varying
by varying ![]() over all densities
containing N electrons. This constraint is introduced by the Lagrange
multiplier,
over all densities
containing N electrons. This constraint is introduced by the Lagrange
multiplier, ![]() , chosen so that
, chosen so that ![]() ,
,
Kohn and Sham chose to separate ![]() into three parts,
so that
into three parts,
so that ![]() becomes
becomes
where ![]() is defined as the kinetic energy
of a non-interacting electron gas with density
is defined as the kinetic energy
of a non-interacting electron gas with density ![]() ,
,
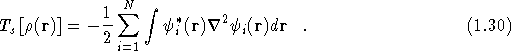
Eq.(![]() ) also acts as a definition for the
exchange-correlation energy functional,
) also acts as a definition for the
exchange-correlation energy functional, ![]() . We
can now rewrite Eq.(
. We
can now rewrite Eq.(![]() ) in terms of an effective potential,
) in terms of an effective potential,
![]() , as follows
, as follows
where
and
![]()
Now, if one considers a system that really contained
non-interacting electrons moving in an external potential equal to
![]() , as defined in Eq.(
, as defined in Eq.(![]() ), then the same
analysis would lead to exactly the same Eq.(
), then the same
analysis would lead to exactly the same Eq.(![]() ). Therefore,
to find the groundstate energy and density,
). Therefore,
to find the groundstate energy and density, ![]() and
and
![]() all one has to do is solve the one-electron
equations
all one has to do is solve the one-electron
equations
As the density is constructed according to
these equations (![]() -
-![]() ) must be solved
self-consistently with Eq.(
) must be solved
self-consistently with Eq.(![]() ).
).
The above derivation assumes that the exchange-correlation functional
is known. At present numerical exchange-correlation potentials have
only been determined for a few simple model systems, and so most
current density functional calculations use the Local Density
Approximation (LDA). The LDA approximates the XC functional
to a simple function of the density at any position, ![]() . The
value of this function is the XC energy per electron in a uniform
homogeneous electron gas of density
. The
value of this function is the XC energy per electron in a uniform
homogeneous electron gas of density ![]() . The LDA expression
for
. The LDA expression
for ![]() is
is
![]()
The LDA is remarkably accurate, but often fails when the electrons are strongly correlated, as in systems containing d and f orbital electrons.