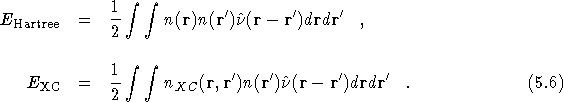In an attempt to understand the origin of these CFSE, it is
instructive to write the electron-electron interaction in terms of a
Hartree interaction, which describes the electrostatic energy of the
system and an Exchange-Correlation interaction, which describes the
interaction of each electron with its exchange-correlation hole, via
an interaction ![]() ;
;
![]()
where
Note the exchange-correlation energy in Eq.(![]() ) is the
full many-body exchange-correlation energy, not the
exchange-correlation energy as defined in density functional theory
(see chapter
) is the
full many-body exchange-correlation energy, not the
exchange-correlation energy as defined in density functional theory
(see chapter ![]() ). It has been established
[3, 74] that both the charge density,
). It has been established
[3, 74] that both the charge density, ![]() and
the shape of the exchange-correlation hole
and
the shape of the exchange-correlation hole ![]()
![]() converge rapidly with
system size and therefore exhibit a very small finite size effect. An
extreme example is jellium (see section
converge rapidly with
system size and therefore exhibit a very small finite size effect. An
extreme example is jellium (see section ![]() ), where the
charge density is exact for all simulation cell sizes but the CFSE is
still present. It therefore appears that the likely source of the
CFSE present in the VMC and HF calculations in figures
), where the
charge density is exact for all simulation cell sizes but the CFSE is
still present. It therefore appears that the likely source of the
CFSE present in the VMC and HF calculations in figures
![]() and
and ![]() is due to the choice of the
interaction,
is due to the choice of the
interaction, ![]() , used in Eq.(
, used in Eq.(![]() ).
In the calculations shown in figures
).
In the calculations shown in figures ![]() and
and
![]() the Ewald interaction has been used to represent the
Coulomb
the Ewald interaction has been used to represent the
Coulomb ![]() interaction between each electron and all the
periodic images of all the other electrons produced by the periodic
boundary conditions. The Ewald interaction has already been described
in detail in chapter
interaction between each electron and all the
periodic images of all the other electrons produced by the periodic
boundary conditions. The Ewald interaction has already been described
in detail in chapter ![]() . Expanding the Ewald
interaction for small r yields
. Expanding the Ewald
interaction for small r yields
where ![]() is the volume of the simulation cell and the tensor,
D, depends on the geometry of the simulation cell (for a cubic
cell D is the identity matrix) and the constant is defined in
chapter
is the volume of the simulation cell and the tensor,
D, depends on the geometry of the simulation cell (for a cubic
cell D is the identity matrix) and the constant is defined in
chapter ![]() so that the average value of the
potential is zero. The deviations from 1/r model the effects of
charges ``outside'' the simulation cell. In the XC integral, however,
the interaction between each electron and its XC hole should be
exactly 1/r, independent of the size of the simulation cell. For
very large simulation cells the 1/r term in the expansion of the
Ewald interaction dominates, but for typical cell sizes such as those
used in figures
so that the average value of the
potential is zero. The deviations from 1/r model the effects of
charges ``outside'' the simulation cell. In the XC integral, however,
the interaction between each electron and its XC hole should be
exactly 1/r, independent of the size of the simulation cell. For
very large simulation cells the 1/r term in the expansion of the
Ewald interaction dominates, but for typical cell sizes such as those
used in figures ![]() and
and
![]() the second term is significant and produces a finite
size error proportional to
the second term is significant and produces a finite
size error proportional to ![]() in the XC energy per electron.
The XC energy is negative and the extra unphysical interaction makes
the XC energy more negative. These observations explain why the HF
energies in figure
in the XC energy per electron.
The XC energy is negative and the extra unphysical interaction makes
the XC energy more negative. These observations explain why the HF
energies in figure ![]() converge with increasing
simulation cell size (i) from below, and (ii) with an error which is
roughly inversely proportional to the number of electrons in the
simulation cell as originally proposed in section
converge with increasing
simulation cell size (i) from below, and (ii) with an error which is
roughly inversely proportional to the number of electrons in the
simulation cell as originally proposed in section ![]() .
.