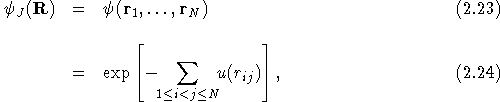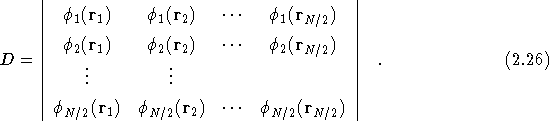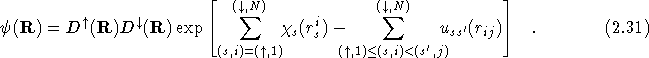To perform a VMC calculation using the algorithm outlined in
figure ![]() , one has to chose the form of the trial
wavefunction,
, one has to chose the form of the trial
wavefunction, ![]() . This trial wavefunction should contain as
much knowledge of the physics of the system being studied as possible.
The choice of
. This trial wavefunction should contain as
much knowledge of the physics of the system being studied as possible.
The choice of ![]() will completely determine the values of all the
observables, such as the energy, obtained from the calculation.
will completely determine the values of all the
observables, such as the energy, obtained from the calculation.
For a bosonic system the many-body wavefunction is a symmetric
function of the coordinates of the particles. McMillan, in his study
of the ground state of liquid ![]() He by the VMC method
[20], used a many-body wavefunction given by a Jastrow
function [21],
He by the VMC method
[20], used a many-body wavefunction given by a Jastrow
function [21],
where u(r) is a two-body function chosen to minimise the energy of
the state. The function, u(r), was chosen so as to enhance the
probability of pairs of ![]() He atoms being separated by a
distance which minimises their interaction energy. The price to be
paid for this is that the kinetic energy is increased due to the
confinement, but the total energy is still reduced.
He atoms being separated by a
distance which minimises their interaction energy. The price to be
paid for this is that the kinetic energy is increased due to the
confinement, but the total energy is still reduced.
For fermionic systems the many-body wavefunction is antisymmetric under particle exchange. The simplest antisymmetric function one can choose is the Slater determinant, often referred to as the Hartree-Fock approximation.
![]()
where
For solids the single particle orbitals, ![]() are normally taken
from either density-functional-theory, local-density-approximation
calculations (DFT-LDA) or Hartree-Fock (HF) calculations. For
all-electron atomic calculations the orbitals used are generally those
obtained from some minimisation scheme [22]. The use of
a separate determinant for up- and down-spin electrons means that the
wavefunction is not antisymmetric on exchange of opposite spin
electrons, however, this form gives the same expectation value as long
as the operator is spin-independent[23]. The advantage of using
two smaller determinants rather than one larger one is that it
is computationally more efficient.
are normally taken
from either density-functional-theory, local-density-approximation
calculations (DFT-LDA) or Hartree-Fock (HF) calculations. For
all-electron atomic calculations the orbitals used are generally those
obtained from some minimisation scheme [22]. The use of
a separate determinant for up- and down-spin electrons means that the
wavefunction is not antisymmetric on exchange of opposite spin
electrons, however, this form gives the same expectation value as long
as the operator is spin-independent[23]. The advantage of using
two smaller determinants rather than one larger one is that it
is computationally more efficient.
In this thesis, we adopt the definition of electron correlation as any
further electron-electron interaction beyond that described by the
exchange interaction in Hartree-Fock theory. According to this
definition, the above form of fermionic wavefunction,
Eq.(![]() ), contains no correlation. In order to introduce
correlation we multiply by a Jastrow factor which is symmetric under
the exchange of particles, giving a wavefunction of the form
), contains no correlation. In order to introduce
correlation we multiply by a Jastrow factor which is symmetric under
the exchange of particles, giving a wavefunction of the form
Two forms of the Jastrow factor are commonly used:
![]()
for solids, and
![]()
for atoms. The ratio of the two parameters (A/F) and the value of a are chosen such that the electron-electron ``cusp'' conditions [24] are obeyed, that is

The value of b can be chosen variationally. For solids the standard
choice for fixing the remaining degree of freedom in the u function
is made by considering the long-range behaviour of u
[25, 26]. More optimised choices for this degree of
freedom are discussed in chapter ![]() . For atoms this
extra degree of freedom is used to either minimise the energy or the
variance of the energy.
. For atoms this
extra degree of freedom is used to either minimise the energy or the
variance of the energy.
Recently more sophisticated Jastrow factors have been used. For atoms [22] this has been done by making the Jastrow factor a function of the electron-nucleus distance as well as the inter-electron distance. Similar schemes have also been implemented for solids [27, 1].
For solids it is found to be beneficial to introduce a one-body,
![]() function which attempts to reverse the effect of the Jastrow
factor on the charge density. As the Jastrow function introduces an
extra repulsion between electrons, this has the effect of smearing out
the charge density. i.e. in regions where the original charge density
is high, the effect of the Jastrow function is to reduce it and
vice-versa. Methods for constructing the
function which attempts to reverse the effect of the Jastrow
factor on the charge density. As the Jastrow function introduces an
extra repulsion between electrons, this has the effect of smearing out
the charge density. i.e. in regions where the original charge density
is high, the effect of the Jastrow function is to reduce it and
vice-versa. Methods for constructing the ![]() function and more
optimised forms of Jastrow function are discussed in
chapter
function and more
optimised forms of Jastrow function are discussed in
chapter ![]() .
.
Thus the final form of the fermionic wavefunction is
This is referred to as the Hartree-Fock-Jastrow-Chi trial wavefunction.