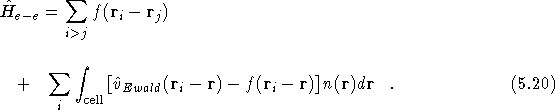Although it would be computationally prohibitive to repeat all the
calculations of figure ![]() within DMC to confirm that
the new interaction behaves in a similar way, it is possible to compare
DMC calculations at the smallest system size, n=2, to see the effect
on the diffusion algorithm of switching to the new electron-electron
interaction.
within DMC to confirm that
the new interaction behaves in a similar way, it is possible to compare
DMC calculations at the smallest system size, n=2, to see the effect
on the diffusion algorithm of switching to the new electron-electron
interaction.
To perform a DMC calculation, one requires not only an energy
expression but also an expression for the Hamiltonian of the system
being studied. The Schrödinger equation may be ``derived'' by
minimising an energy functional, ![]() , where
, where ![]() is a normalised wavefunction. If a
similar procedure is carried out for a functional including the
electron-electron interaction of Eq. (
is a normalised wavefunction. If a
similar procedure is carried out for a functional including the
electron-electron interaction of Eq. (![]() ), the
electron-electron interaction operator in the resulting
Schrödinger-like equation is
), the
electron-electron interaction operator in the resulting
Schrödinger-like equation is
Again, as in Eq. (![]() ), we chose to use the LDA
charge density,
), we chose to use the LDA
charge density, ![]() , as the input density,
, as the input density, ![]() , to the second term in the Hamiltonian. The total
electron-electron energy,
, to the second term in the Hamiltonian. The total
electron-electron energy, ![]() , is then the expectation value of
, is then the expectation value of
![]() minus a double counting term for the electrostatic
interactions;
minus a double counting term for the electrostatic
interactions;
This double counting term can itself be accumulated during the DMC calculation and then subtracted off at the end of the simulation as it is a fixed constant that will not affect the diffusion algorithm.
Two DMC calculations were performed on the n=2 system of
diamond-structure silicon to compare the effect of the new interaction
in DMC and VMC. The DMC calculations were performed using a Slater
determinant of single-particle orbitals with ![]() -points chosen
on a reciprocal space grid centred at the origin, i.e.
-points chosen
on a reciprocal space grid centred at the origin, i.e. ![]() -point
sampling. This sampling was chosen as DMC calculations with
-point
sampling. This sampling was chosen as DMC calculations with
![]() -point sampling are required in the next chapter as well.
-point sampling are required in the next chapter as well.
The first DMC calculation was performed with the standard Hamiltonian
as described in chapter ![]() . The second
calculation used the new DMC Hamiltonian from
Eq.(
. The second
calculation used the new DMC Hamiltonian from
Eq.(![]() ). The same changes were made to the DMC
algorithm (described in chapter
). The same changes were made to the DMC
algorithm (described in chapter ![]() ) as were
made to the VMC algorithm in section
) as were
made to the VMC algorithm in section ![]() to implement the
new electron-electron interaction. The new DMC calculation exhibited
the same stability in the population of walkers as the original
Hamiltonian. It required a similar number of steps to diffuse to a
state where energies could be accumulated and the intrinsic variance
of the energy over the run was also very similar. Table
to implement the
new electron-electron interaction. The new DMC calculation exhibited
the same stability in the population of walkers as the original
Hamiltonian. It required a similar number of steps to diffuse to a
state where energies could be accumulated and the intrinsic variance
of the energy over the run was also very similar. Table ![]() shows a comparison of the total energies obtained in VMC and DMC using
the Ewald and new electron-electron interactions. The VMC results in
table
shows a comparison of the total energies obtained in VMC and DMC using
the Ewald and new electron-electron interactions. The VMC results in
table ![]() were performed using
were performed using ![]() -point sampling
to facilitate the comparison.
-point sampling
to facilitate the comparison.
| Ewald Interaction | New Interaction | |
| (eV per atom) | (eV per atom) | |
| VMC | -106.88 | -106.70 |
| DMC | -107.41 | -107.30 |
|
| 0.53 | 0.60 |
The results show that the reduction in energy obtained by performing a DMC calculation rather than a VMC calculation is similar for the two electron-electron interactions. This suggests that, as expected, the finite size effects in DMC broadly follow those in VMC and that using the new electron-electron interaction yields a similar improvement in DMC calculations to VMC calculations.