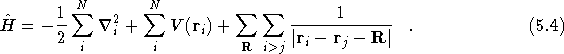In an attempt to gain further understanding on the nature of these
CFSE, the VMC calculations shown in figure ![]() , were
repeated using the Local Density Approximation (LDA) to Density
Functional Theory (DFT) and Hartree-Fock (HF) theory. The k-point
sampling in the LDA and HF calculations was chosen to be consistent
with choosing simulation cell sizes of n= 2,3,4 and 5 as in the VMC
calculations. The results of these LDA and HF calculations are
plotted in figures
, were
repeated using the Local Density Approximation (LDA) to Density
Functional Theory (DFT) and Hartree-Fock (HF) theory. The k-point
sampling in the LDA and HF calculations was chosen to be consistent
with choosing simulation cell sizes of n= 2,3,4 and 5 as in the VMC
calculations. The results of these LDA and HF calculations are
plotted in figures ![]() and
and ![]() . Note the
scale of the y-axis is the same in both graphs.
. Note the
scale of the y-axis is the same in both graphs.
Figure: Total energy per atom calculated using LDA as a function of system size.
Figure: Total energy per atom calculated using HF, as a function of system size.
To facilitate comparison between the LDA and HF results the LDA
orbitals were used to calculate the HF energies, so that the energy
differences arise solely from the difference between the LDA
exchange-correlation (XC) energy and the HF exchange energy.
Figure ![]() shows that the LDA energy converges very
rapidly with simulation cell size, whereas in figure
shows that the LDA energy converges very
rapidly with simulation cell size, whereas in figure
![]() , the HF exchange energy converges very slowly with
simulation cell size as did the VMC in figure
, the HF exchange energy converges very slowly with
simulation cell size as did the VMC in figure ![]() . For
n=3 the finite size error in the LDA energy (IPFSE) is 0.012 eV per
atom, which is much smaller than the HF finite size error of -0.211 eV
per atom. The slow convergence of the HF exchange energy with the
density of BZ sampling (which is equivalent to the size of the
simulation cell) is well known[71, 72, 73]
and is usually solved by increasing the quality of the BZ integration.
. For
n=3 the finite size error in the LDA energy (IPFSE) is 0.012 eV per
atom, which is much smaller than the HF finite size error of -0.211 eV
per atom. The slow convergence of the HF exchange energy with the
density of BZ sampling (which is equivalent to the size of the
simulation cell) is well known[71, 72, 73]
and is usually solved by increasing the quality of the BZ integration.
The rapid convergence of the LDA energy with simulation cell size is easily understood. In the LDA, the total energy can be written as a functional of the charge density
The LDA charge density ![]() has been shown [3] to
converge rapidly with simulation cell size, hence the total energy also
converges rapidly. As the LDA orbitals were used to calculate the HF
energies, the kinetic and external potential energy in the LDA and HF
calculations in figures
has been shown [3] to
converge rapidly with simulation cell size, hence the total energy also
converges rapidly. As the LDA orbitals were used to calculate the HF
energies, the kinetic and external potential energy in the LDA and HF
calculations in figures ![]() and
and ![]() are
identical for each system size. If one considers the Hamiltonian
for the system as in Eq.(
are
identical for each system size. If one considers the Hamiltonian
for the system as in Eq.(![]() ), the expectation value of
the first and second terms of the Hamiltonian with respect to the
trial wavefunction are the same in the LDA and HF and only differ in
the final electron-electron interaction term;
), the expectation value of
the first and second terms of the Hamiltonian with respect to the
trial wavefunction are the same in the LDA and HF and only differ in
the final electron-electron interaction term;
Hence, the original interpretation of the the residual finite size effect after the subtraction of the IPFSE as a Coulomb FSE is consistent with these results.