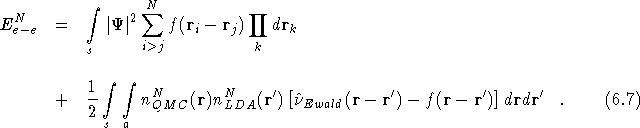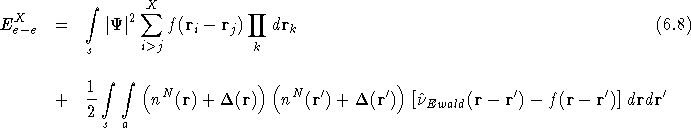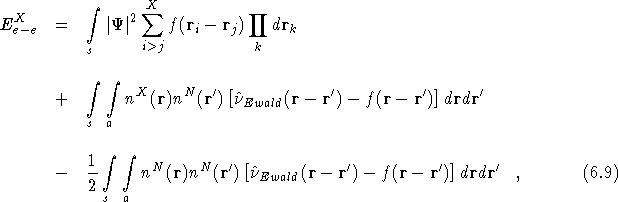The expression for the gap energy in Eq.(![]() ) is based on
the change in the ground state energy of the bulk solid when a single
electron is added or removed. In all our QMC calculations for bulk
solids, we choose to model this system using a finite simulation cell
to which we apply periodic boundary conditions. When an additional
electron is added to this simulation cell, the periodic boundary
conditions effectively introduce an equivalent extra electron into
each of the periodic images of the simulation cell. This adds an
extra electrostatic energy into the system, which when combined with a
compensating background charge is just the Madelung energy of an
electron (hole) crystal with the periodicity of the simulation cell.
A similar effect has been observed in LDA
calculations[105, 106, 107, 108]. Leslie and
Gillan[109] proposed a correction term to the Hartree
energy of the system to account for the additional electrostatic
energy due to an array of charged defects,
) is based on
the change in the ground state energy of the bulk solid when a single
electron is added or removed. In all our QMC calculations for bulk
solids, we choose to model this system using a finite simulation cell
to which we apply periodic boundary conditions. When an additional
electron is added to this simulation cell, the periodic boundary
conditions effectively introduce an equivalent extra electron into
each of the periodic images of the simulation cell. This adds an
extra electrostatic energy into the system, which when combined with a
compensating background charge is just the Madelung energy of an
electron (hole) crystal with the periodicity of the simulation cell.
A similar effect has been observed in LDA
calculations[105, 106, 107, 108]. Leslie and
Gillan[109] proposed a correction term to the Hartree
energy of the system to account for the additional electrostatic
energy due to an array of charged defects,
![]()
where ![]() is the Madelung constant for the supercell
geometry, L is the length of the simulation cell,
is the Madelung constant for the supercell
geometry, L is the length of the simulation cell, ![]() is
a dielectric constant for the material, and q is the charge on the
defect. The problem with this correction is how to choose the
dielectric constant,
is
a dielectric constant for the material, and q is the charge on the
defect. The problem with this correction is how to choose the
dielectric constant, ![]() . In general experimental values
have been used and these have not been found to work particularly
well.
. In general experimental values
have been used and these have not been found to work particularly
well.
It has also been speculated by Engel et al.[79] that similar effects may be present in their VMC calculations of the band structure of a two-dimensional model crystal. In their calculations, an extra electron was added into an orbital in the conduction band. However, this orbital is actually spread throughout the simulation cell and so can be regarded as contributing a much smaller term to the Hartree energy than a point defect plus background would. In the limit of infinite simulation cell size L, any additional energy due to interactions between the array of additional electrons would disappear. Therefore Engel et al. treat this as a finite size effect and deal with it by fitting results for a series of VMC calculations at different system sizes to the expression
where L is the length of the simulation cell, and
![]() is a parameter that represents a reduced
is a parameter that represents a reduced ![]() due
to the screening of the other valence electrons.
due
to the screening of the other valence electrons.
In our QMC calculations[110, 3, 111] we no longer use the Ewald interaction to evaluate the electron-electron interaction between pairs of electrons and therefore we are not necessarily restricted to including all the periodic images of the additional electron(hole) in our system in the same way as Engel et al.
Figure: Addition of a single electron to the simulation cell. Figure
(a) shows an N electron simulation cell periodically repeated. Figure
(b) shows the same bulk system with an additional electron added only
to the simulation cell (red).
Consider the two systems illustrated in figure ![]() .
Figure (a) schematically represents the standard simulation cell for
the N electron system and a few of the periodic images of the
simulation cell. The electron-electron energy associated with this
system can be defined as in section
.
Figure (a) schematically represents the standard simulation cell for
the N electron system and a few of the periodic images of the
simulation cell. The electron-electron energy associated with this
system can be defined as in section ![]() by the new
electron-electron energy expression for N electron systems,
by the new
electron-electron energy expression for N electron systems,
In Figure ![]() (b) the same system is shown with an extra
electron added only to the actual simulation cell, not to any of
its periodic images. We can represent the change in the charge
density of the whole system due to the additional electron by
(b) the same system is shown with an extra
electron added only to the actual simulation cell, not to any of
its periodic images. We can represent the change in the charge
density of the whole system due to the additional electron by
![]() , and we would like to confine
, and we would like to confine ![]() to
within the central simulation cell, i.e. there should be no additional
electrons in the periodic images of the simulation cell. This effect
can be achieved by altering the interaction so that each electron
`feels' the full 1/r interaction with all N+1 electrons within the
simulation cell surrounding it
to
within the central simulation cell, i.e. there should be no additional
electrons in the periodic images of the simulation cell. This effect
can be achieved by altering the interaction so that each electron
`feels' the full 1/r interaction with all N+1 electrons within the
simulation cell surrounding it![]() , but only feels the Hartree
interaction with the charge density due to N electrons in each
periodic image outside the simulation cell. We re-write
Eq.(
, but only feels the Hartree
interaction with the charge density due to N electrons in each
periodic image outside the simulation cell. We re-write
Eq.(![]() ) to take account of the extra electron and the
change in the charge density,
) to take account of the extra electron and the
change in the charge density, ![]() , which is confined to
the central simulation cell, as
, which is confined to
the central simulation cell, as
and expand out the product
![]() ,
in the second term. We can discard the term in
,
in the second term. We can discard the term in
![]() which is small as
which is small as ![]() is a short ranged function and
is a short ranged function and
![]() is
a small for
is
a small for ![]() small. This can be understood
physically in the following way;
small. This can be understood
physically in the following way; ![]() represents the
change in the charge density due to adding an electron. The
represents the
change in the charge density due to adding an electron. The
![]() term represents the interaction of
this change with itself. In an infinite system
term represents the interaction of
this change with itself. In an infinite system ![]() is
virtually zero and so this term should disappear. Removing this term yields
is
virtually zero and so this term should disappear. Removing this term yields
where ![]() .
.
As in Eq.(![]() ), the first term describes the full
Hartree and exchange/correlation interaction between all X electrons
in the simulation cell. The second two terms can be interpreted as
representing the Hartree interaction between the charge density due to
X electrons inside the simulation cell and the charge density due to
N electrons outside the simulation cell. Therefore, as far as the
electron-electron interaction is concerned there is only one
extra electron present in the system rather than the whole periodic
array which is normally introduced. The use of this new energy
expression removes the need for ad hoc corrections to the finite
size effects such as those used by Engel et al. in
Eq.(
), the first term describes the full
Hartree and exchange/correlation interaction between all X electrons
in the simulation cell. The second two terms can be interpreted as
representing the Hartree interaction between the charge density due to
X electrons inside the simulation cell and the charge density due to
N electrons outside the simulation cell. Therefore, as far as the
electron-electron interaction is concerned there is only one
extra electron present in the system rather than the whole periodic
array which is normally introduced. The use of this new energy
expression removes the need for ad hoc corrections to the finite
size effects such as those used by Engel et al. in
Eq.(![]() ). Note, when using either of the two
energy expressions, Eq.(
). Note, when using either of the two
energy expressions, Eq.(![]() ) and
Eq.(
) and
Eq.(![]() ), we include background charges so there is no
contribution to the total energy or any gap energies from the
), we include background charges so there is no
contribution to the total energy or any gap energies from the
![]() component of the f interaction. This is equivalent to
ensuring that each cell is neutral, as would be the case when a single
electron is added to the infinite system.
component of the f interaction. This is equivalent to
ensuring that each cell is neutral, as would be the case when a single
electron is added to the infinite system.