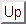

Next: References
Up: Geometry Optimisation with CASTEP
Previous: A Variable Cell calculation
When the supercell does not have full 3D periodicity the calculation must be converged with respect to other supercell
parameters. Consider a surface. We will use the Si(100) surface reconstruction as an example. Figure 5 below shows
the bulk terminated Si(100) surface.
Figure 5:
The bulk terminated Si(100) surface
|
|
In one direction we have a defect; after some point there will be a complete absence of atoms, essentially out to infinity.
In the opposite direction we move into the bulk and in principle we should include an infinite number of bulk layers below
the top few surface layers.
Both of these requirements are computationally impractical. In practice we must include just enough bulk layers
so that the inner bulk atoms remain in place during the calculation and only the top surface layers reconstruct.Also,
only a finite amount of vacuum space above the top surface layer is practical. However, when a supercell is repeated
periodically in all three spatial directions the bottom bulk-like region of one supercell borders upon the top vacuum
region of the supercell below it (figure 6). If the vacuum region is not thick enough the uncompensated charges on the
bottom of the one supercell and the top of the supercell under it will cause an unphysical interaction. Vertically
adjacent supercells must not be able to 'see' one another.
Figure 6:
A finite vacuum gap may cause an interaction between neighbouring atomic layers in vertically adjacent
supercells
|
|
Other effects are caused by uncompensated charges. The bottom layer of the supercell, which should be bulk-like, may
reconstruct. To prevent this we can constrain these atoms to remain in their bulk positions. Another effect is an
interaction between the top surface layer uncompensated charges and the bottom layer uncompensated charges through the
intermediate layers. The supercell must contain sufficient numbers of layers of atoms to space the top and bottom layers
out so as to minimise the internal interactions in the supercell. A neat trick is to use hydrogen passivation. In this
the dangling bonds of the bottom bulk like layers are compensated by bonding these atoms to hydrogen atoms. This
introduces extra atoms into the calculation but reduces the overall number of atoms needed because the number of spacer
layers in the supercell is reduced (also, hydrogen is computationally cheap). The thickness of the vacuum gap can also
be reduced when hydrogen passivation is used (remember empty space is a computational expense too).
The Si(100) surface is known to have a simple 2x1 dimerised structure. The small in-plane unit cell of the reconstructed
surface means that we can use a small supercell and we do not need many atoms. The following initial supercell will be
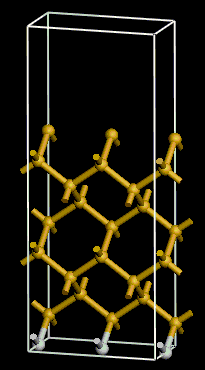
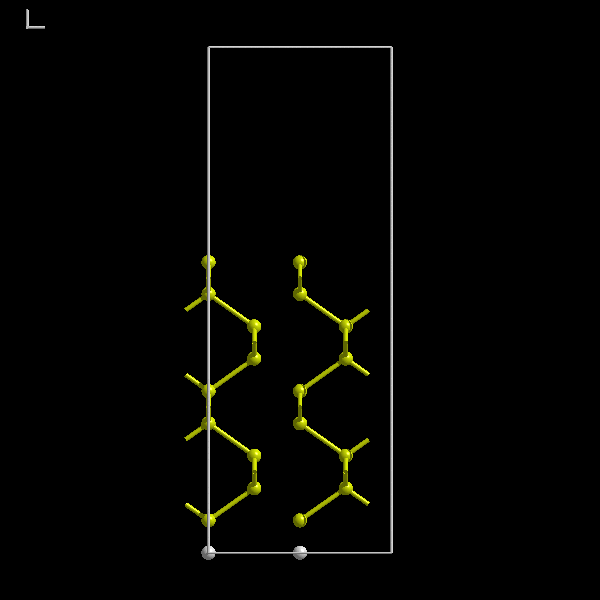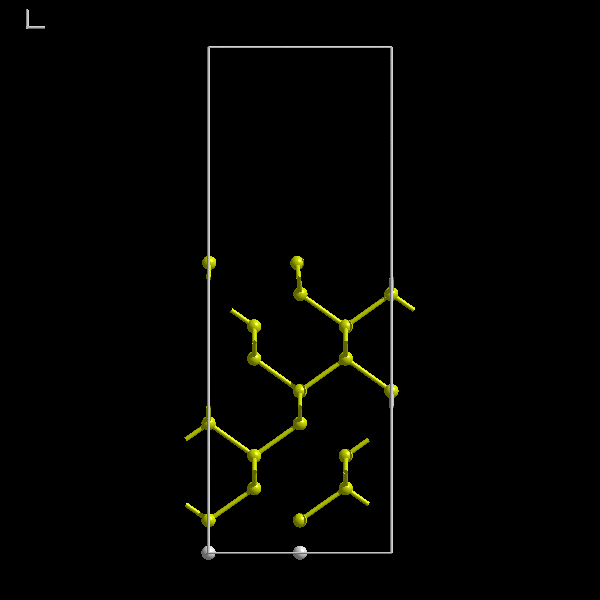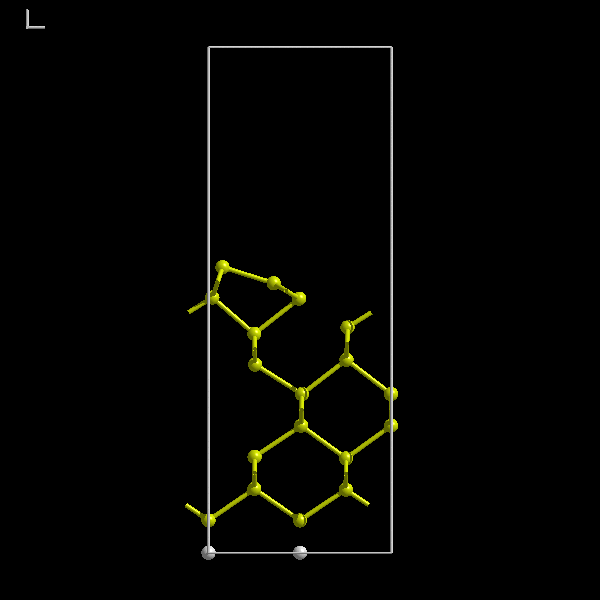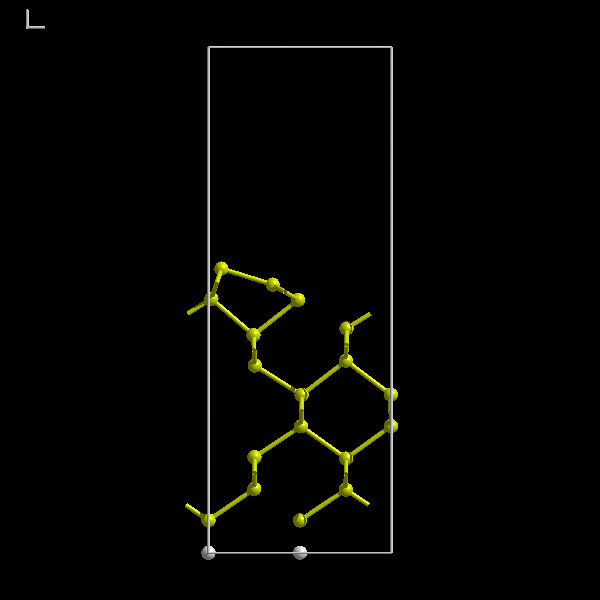
sufficient (figure 7). Notice the hydrogen passivation.
Figure 7:
A hydrogen passivated Si(100) supercell with a 7 Å vacuum gap and 9 layers of silicon.
|
|
This is a crucial feature of performing geometry optimisation on surfaces. The initial surface unit cell must be large
enough to enclose any expected reconstruction. A Si(111)-1x1 supercell will not reconstruct into the Si(111)-7x7
Takayanagi reconstruction! If no suggestions have been provided as to possible reconstructed periodicities by
previous theoretical or experimental work then the user might have to begin with a large supercell and perform a
variable cell calculation (in case of lateral relaxations).
Now we must perform the abovementioned supercell convergence checks. We will use a 9 k-point MP grid with a cutoff
energy of 260eV for this (we will converge our calculation with respect to basis set parameters later). Let us start
with a large vacuum gap of 15Å (I know that this is entirely sufficient from experience). We will now be able to
perform several calculations with 7, 8, 9, 10 and 11 layers of silicon and not have to worry about the vacuum gap - one
problem at a time. The supercell lattice matrix and the atomic coordinate list in the .cell file for 7 layers of silicon
looks like
%BLOCK LATTICE_CART
7.6801695932 0.0000000000 0.0000000000
0.0000000000 3.8801700000 0.0000000000
0.0000000000 0.0000000000 24.5037250000
%ENDBLOCK LATTICE_CART
%BLOCK POSITIONS_FRAC
H 0.2500000000 -0.0103307854 -0.0000000000
H 0.7500000000 -0.0103307854 -0.0000000000
Si 0.5000000000 0.9896692146 0.1662206460
Si 0.5000000000 0.9896692146 0.3878481741
Si 0.2500000000 0.4948346073 0.0554068820
Si 0.2500000000 0.4948346073 0.2770344101
Si 0.7500000000 0.4948346073 0.0554068820
Si 0.7500000000 0.4948346073 0.2770344101
Si 0.0000000000 0.9896692146 0.1662206460
Si 0.0000000000 0.9896692146 0.3878481741
Si -0.0000000000 0.4948346073 0.1108137640
Si -0.0000000000 0.4948346073 0.3324412921
Si 0.7500000000 0.9896692146 0.2216275281
Si 0.5000000000 0.4948346073 0.1108137640
Si 0.5000000000 0.4948346073 0.3324412921
Si 0.2500000000 0.9896692146 0.2216275281
%ENDBLOCK POSITIONS_FRAC
Notice the hydrogen atoms. Now for the constraints on the bottom layer silicon atoms and the hydrogen atoms (the H atoms
must also be constrained because the have a tendency to move into awkward formations). The list of constraints whose
format was specified before looks like
%BLOCK IONIC_CONSTRAINTS
1 H 1 1.00000000 0.00000000 0.00000000
2 H 1 0.00000000 1.00000000 0.00000000
3 H 1 0.00000000 0.00000000 1.00000000
4 H 2 1.00000000 0.00000000 0.00000000
5 H 2 0.00000000 1.00000000 0.00000000
6 H 2 0.00000000 0.00000000 1.00000000
7 Si 3 1.00000000 0.00000000 0.00000000
8 Si 3 0.00000000 1.00000000 0.00000000
9 Si 3 0.00000000 0.00000000 1.00000000
10 Si 5 1.00000000 0.00000000 0.00000000
11 Si 5 0.00000000 1.00000000 0.00000000
12 Si 5 0.00000000 0.00000000 1.00000000
%ENDBLOCK IONIC_CONSTRAINTS
NB You will also need to add some other settings to your cell file, especially
fix_com = FALSE
fix_all_cell = TRUE
fix_all_ions = FALSE
as otherwise these constraints will interfere. See the ionic constraints section for more details.
So now we have our supercells ready to run. The calculations were performed on 9 nodes of a Beowulf cluster. The total
final energy per supercell atom as a function of the number of layers of silicon is shown in figure 8 below
Figure 8:
The total final energy per supercell atom as a function of the number of layers of silicon.
|
|
We can see that the energy per atom is decreasing almost linearly with the number of layers. We would need to add very
many layers for the energy to not vary by the inclusion of more layers. We will use 9 layers of silicon in future
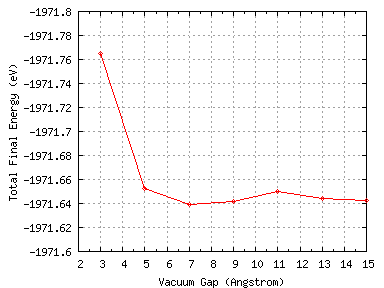
calculations. With 9 silicon layers the vacuum gap was varied from 3 Å to 15 Å in 2 Å steps. The total final
energy as a function of vacuum gap separation is shown below.
Figure 9:
The total final energy as a function of the vacuum gap thickness
|
|
We can see that the total energy is converged with respect to the vacuum gap when this has a thickness greater than
about 7 Å (there is a slight upwards kink after this). We will use a 9 Å vacuum gap in all future
calculations.
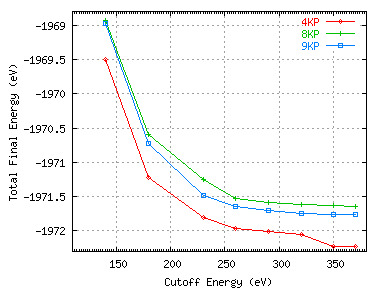
Now it remains to converge the basis set paramters. Cutoff energies of 140, 180, 230, 260, 290, 320, 350, and 370eV were
used with MP grids containing 4, 8 and 9 k-points. The calculations were performed on a Beowulf cluster using 8 nodes
for the 4 and 8 k-point calculations and 9 nodes for the 9 k-point calculation. The convergence of the total energy
with basis set parameters is shown below.
Figure 10:
Convergence of the total energy with respect to the basis set parameters
|
|
We can see that using 9 layers of silicon, a 9 Å vacuum gap, 9 k-points and a 370 eV cutoff energy will ensure
minimisation of systematic errors with respect to both supercell aperiodicity and the basis set repectively.
So what results do we get? Click on the movie below to see the evolution of the atomic positions as the calculation
progresses. Please use the 'back' button to close the movie.
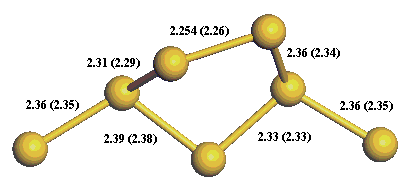
We can see that the result is an initial vertical relaxation followed by asymmetric dimerisation of the top layer silicon
atoms. Asymmetric dimerisation has been seen experimentally [6] and has been confirmed by detailed theoretical
calculations [7]. The bond lengths are shown in figure 11 below (the numbers in brackets are taken from reference [7]).
Figure 11:
Principle bond lengths compared with the results of other theoretical work (in brackets).
|
|
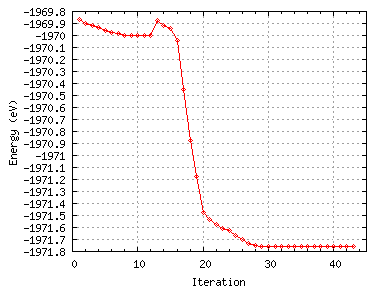
It is interesting to look at the evolution of the total energy and the forces as the calculation progresses. Figures 12
and 13 below shows this.
Figure 12:
The variation of the total energy with BFGS iteration number.
|
|
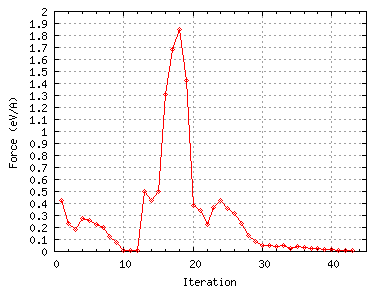
Figure 13:
The variation of the maximum force with BFGS iteration number.
|
|
There is an inital dip in the forces at around iteration 14 as some search direction is tried but this causes an
increase in the energy. Then there is a large increase in the forces (presumably due due dimer attraction)
accompanied by a large energy gain which are due to dimerisation. There is then a small peak in the forces and the
energy at about iteration 24 as the energy barrier to asymmetric dimerisation is overcome.
In summary, this example has illustrated the use of constraints upon atomic positions and has also looked at hydogen
passivation and the variation of both the number of layers of atoms and the thickness of the vacuum gap as ways to
deal with aperiodic supercell configurations such as surfaces.
Next: References
Up: Geometry Optimisation with CASTEP
Previous: A Variable Cell calculation
2005-04-03
![\includegraphics[scale=0.5]{vg.eps}](img50.gif)
![\includegraphics[scale=0.5]{bulk_si100.eps}](img49.gif)