Joseph Nelson

Dr Joseph Nelson
Member of Churchill College
Postdoc in Dr Morris's group
Office: 514 Mott Bld
Phone: +44(0)1223 3 37278
Email: jn336 @ cam.ac.uk
ORCID: 0000-0002-5653-092X
Google scholar page
TCM Group, Cavendish Laboratory
19 JJ Thomson Avenue,
Cambridge, CB3 0HE UK.
Research
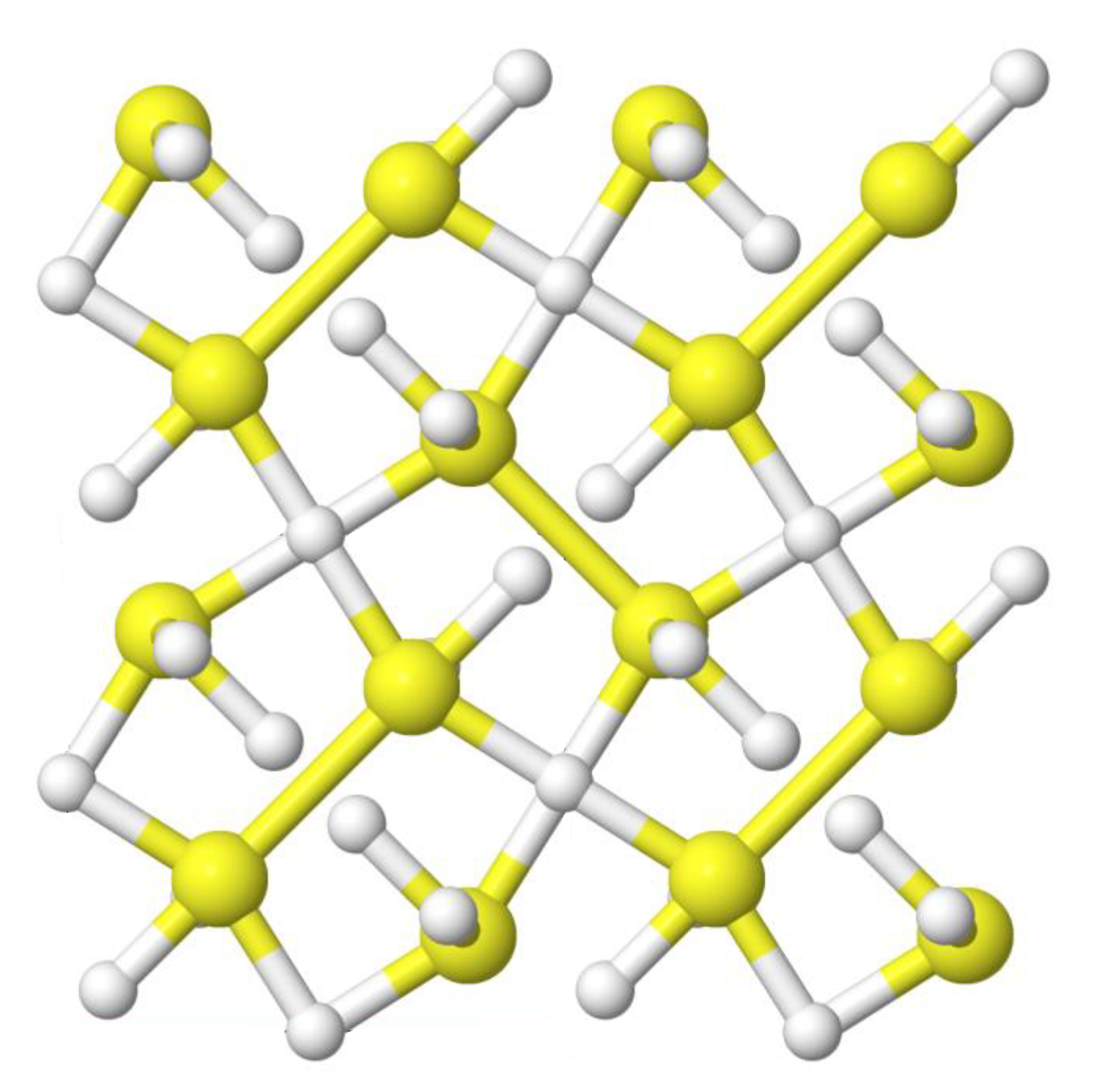
I use the ab initio random structure searching (AIRSS) technique to predict the crystal structures formed by compounds, with a particular focus on high-pressure phases (of the order of GPa). AIRSS generates candidate structures stochastically, and alongside density-functional theory (DFT) calculations, which I typically perform using the CASTEP plane-wave pseudopotential DFT code, allows the efficient prediction and characterisation of crystal structures.
I've recently worked on the prediction of stable compounds formed by hydrogen and sulfur at pressures up to 300 GPa. Record-high temperature superconductivity has recently been reported in compressed samples of H2S, with a critical temperature of 203 K observed at pressures near 155 GPa. A complete explanation of this behaviour requires us to understand the stable crystal structures formed under these conditions.
In Plain English
The properties of a solid depend intimately on the exact arrangement of its consistuent atoms, and the strength of chemical bonding between those atoms. A very familiar example is carbon, where atoms can be arranged in 2D hexagonal sheets, creating a soft, flaky and opaque material (graphite), or with atoms in a 3D network, creating an extremely hard and transparent material (diamond). My research uses methods that allow us to predict the arrangement of atoms in a solid (its crystal structure) from first principles - that is, without recourse to any prior knowledge, other than the constituent atoms.
We can get some insight into the structure and properties of a solid by using basic chemistry. This supplies us with rules about the number and nature of chemical bonds that atoms form, based on their position in the periodic table - e.g., carbon likes to surround itself with four covalent bonds, oxygen two, and so on. However, a full and accurate description of the chemical bonding in a solid requires the laws of quantum mechanics. Given a particular crystal structure, my research uses a branch of quantum mechanics called density functional theory, which allows us to calculate how electrons are distributed in the structure. From this, we gain insights into the types of chemical bonding occuring in a solid, and we are also able to calculate a plethora of other properties such as mechanical strength, optical properties and electrical conductivity. I'm particularly interested in the behaviour of solids at very high pressures (such as those found at the center of the Earth), where unexpected behaviour can emerge and the rules of basic chemistry don't always apply.
Featured Publications
-
High-pressure CaF2 revisited: a new high-temperature phase and the role of phonons in the search for superionic conductivity
Phys. Rev. B 98, 224105 (2018) [arXiv:1811.08740] -
Sodiation and Desodiation via Helical Phosphorus Intermediates in High-Capacity Anodes for Sodium-Ion Batteries
J. Am. Chem. Soc. 140, 7994-8004 (2018) -
High-pressure phases of group-II difluorides: Polymorphism and superionicity
Paper: Phys. Rev. B 95, 054118 (2017) [arXiv:1702.00746]; Reply: Phys. Rev. B 98, 186102 (2018) [arXiv:1811.08745] -
Quantum hydrogen-bond symmetrization in the superconducting hydrogen sulfide system
Nature 532, 81-84 (2016) [arXiv:1512.02933] -
Dissociation products and structures of solid H2S at strong compression
Editor's Suggestion, Phys. Rev. B 93, 020103(R) (2016) [arXiv:1508.03900] -
High-pressure hydrogen sulfide from first principles: a strongly anharmonic phonon-mediated superconductor
Editor's Suggestion, Phys. Rev. Lett. 114, 157004 (2015) [arXiv:1502.02832] -
Calcium peroxide from ambient to high pressures
Phys. Chem. Chem. Phys. 17, 6889-6895 (2015) [arXiv:1502.03329]