Predicting the core level spectra of BN from first principles
Purpose: Introduces the core level spectroscopy feature in CASTEP.
Modules: Materials Visualizer, CASTEP
Time: 
Prerequisites: Using the crystal builder
Background
Density functional theory provides a robust and reliable framework for calculations of core-level spectra. This technique has been applied to numerous systems with good results (Gao et al., 2008). DFT as a method is somewhat limited, and recent developments in core-level spectroscopy calculations using the Bethe-Salpeter equation or time-dependent DFT (or a combination of the two) provide more rigorous approaches for a core level spectrum calculation. In these methods many-body effects, such as the broadening of spectra due to electron-hole lifetime, can be taken into account. While good agreement with experimental results has been achieved, this necessarily comes with a much greater computational burden in comparison to standard ground state DFT calculations.
A systematic observation of core hole effects and a quantitative estimation of the core hole strength in materials can assist in the simulation and interpretation of core level spectra. Applying empirical rules, a core-level spectrum is not likely to be heavily influenced by core-hole effects, so information on the ground electronic structure of a material can be inferred directly from the experimental spectrum. In this case, traditional ground state calculations should predict the main features of experimental results and allow interpretation of the spectrum. If this is not the case, inclusion of the influence of the core hole is essential in the theoretical simulation. Analysis of experimental results should take into account that near-edge fine structure includes influences in the core excitation processes beyond the ground state electronic structure.
For a more general discussion of the core level spectroscopy and details of its implementation see S.-P. Gao et al., 2008 and references.
Introduction
CASTEP can calculate spectroscopic properties of solids that are due to electronic transitions from a core level of an ion to the conduction band (X-ray absorption) and from the valence band to a core level (X-ray emission). This can be used to describe a wide variety of experimental results connected to such processes. Core holes can be created by X-ray or electron incident radiation.
The core level is localized, so core level spectroscopy provides a detailed element-specific picture of the local electronic structure around a given atomic site. There is no contribution from the other atoms in the system, so the electronic states for a specific atom can be investigated.
In the case of anisotropic systems angular-dependent experiments enable the separation of states with different symmetries for the involved orbitals. An important consequence is that symmetry states which result solely from chemical bonding can be studied. For further information see the core level spectroscopy theory topic.
This tutorial illustrates how CASTEP can be used to determine the core level spectra of a material in Materials Studio using quantum mechanical methods. You will learn how to build a crystal structure and set up a CASTEP energy calculation, followed by core-hole spectroscopy calculations and then analyze the results.
This tutorial covers:
- Getting started
- To set up and run a CASTEP calculation
- To set up the core holes
- To set up and run a CASTEP calculation with core holes
- To analyze the results
- To compare the results with experimental data
In order to ensure that you can follow this tutorial exactly as intended, you should use the Settings Organizer dialog to ensure that all your project settings are set to their BIOVIA default values. See the Creating a project tutorial for instructions on how to restore default project settings.
Begin by starting Materials Studio and creating a new project.
Open the New Project dialog and enter BN as the project name, click the OK button.
The new project is created with BN listed in the Project Explorer. Now you will import the BN structure for your calculation.
Select File | Import... from the menu bar to open the Import Document dialog. Navigate to the folder Structures/semiconductors/ and select BN.xsd. Click the Open button.
The crystal structure in the 3D Viewer is the conventional unit cell containing 8 atoms, which shows the cubic symmetry of the lattice. CASTEP uses the full symmetry of the lattice if any exists, so the primitive lattice, containing 2 atoms per unit cell, can be used. The charge density, bond distances, and total energy per atom will be the same no matter how the unit cell is defined, so by using fewer atoms in the unit cell, the computation time is decreased.
The only time that care is needed is when a spin-polarized calculation is performed on a magnetic system, where the charge density spin wave has a period which is a multiple of the primitive unit cell.
Choose Build | Symmetry | Primitive Cell from the menu bar.
The 3D Viewer displays the primitive cell.
BN primitive cell
2. To set up and run a CASTEP calculation
Click the CASTEP button  on the Modules toolbar and select Calculation from the dropdown list or choose
Modules | CASTEP | Calculation from the menu bar.
on the Modules toolbar and select Calculation from the dropdown list or choose
Modules | CASTEP | Calculation from the menu bar.
This opens the CASTEP Calculation dialog.
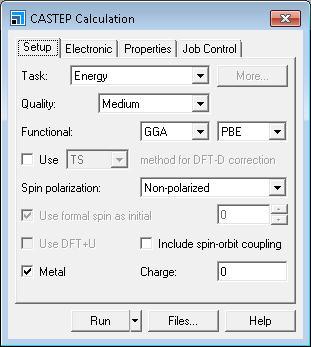
CASTEP Calculation dialog, Setup tab
On the Setup tab of the CASTEP Calculation dialog, ensure the Task is set to Energy and the Quality is set to Medium.
If you change the quality, the other parameters change to reflect this. Next, you will specify the electronic options for the calculation.
On the Electronic tab, select OTFG ultrasoft from the Pseudopotentials dropdown list.
On the fly (OTFG) pseudopotentials are required to compute the core level spectra. If you choose a different type of pseudopotential, the core level spectroscopy calculation cannot be performed.
Next, you will specify which properties you want to calculate.
On the Properties tab, check the Core level spectroscopy checkbox and set the Energy range to 40 eV.
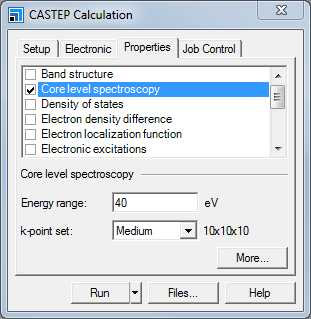
CASTEP Calculation dialog, Properties tab
An energy range of 40 eV for the core level spectroscopy will allow access to energies up to 40 eV above the Fermi level.
On the Job Control tab click the More... button to open the CASTEP Job Control Options dialog. Change the Update interval to 30.0 s and close the dialog.
If you wish to run the calculation on a remote server, you can specify this on the Job Control tab.
Click the Run button and close the dialog.
After a few seconds, a new folder is displayed in the Project Explorer and this will contain all the results from the calculation. The Job Explorer is displayed, which contains information about the status of the job.
When the job finishes, the files are transferred back to the client and this can take some time due to the size of certain files. When the results are transferred, you should have several documents, among them:
BN.xsd- the crystal structureBN.castep- an output file from the CASTEP energy calculationBN_EELS.castep- an output file from the CASTEP core level spectroscopy calculationBN.paramandBN_EELS.param- input information, saving the input files before proceeding with the calculation creates these files and gives you the opportunity to edit them
You will find these files in the Project Explorer in a folder called BN CASTEP Energy once the calculation completes.
To simulate core hole effects you should create a supercell of sufficiently large size, so that artificial interactions between periodic images of the atoms containing core holes are reduced. However, large supercells are very computationally expensive, so a supercell of 32 atoms (6 Å) is used to strike a balance in this tutorial.
For investigative experiments, where greater computational resources are available, larger supercells should be used in order to obtain more accurate results.
Now you will construct a supercell from the primitive cell by redefining the extent of the lattice.
Make BN.xsd in the project root the active document.
Select Build | Symmetry | Redefine Lattice from the menu bar to open the Redefine Lattice dialog.
Enter -3 1 1 for A, -1 -1 3 for B,
and 1 1 1 for C. Click the Redefine button and close the dialog.
Select File| Save As... from the menu bar and save the 3D Atomistic document as BN_N_hole.xsd. Repeat
this to save the document as BN_B_hole.xsd.
Choose Window | Close All from the menu bar, then double-click on BN_N_hole.xsd in the Project Explorer.
You will create a core hole on a nitrogen atom and ensure that the symmetry is properly imposed.
Select any N atom in the supercell.
Select Modify | Electronic Configuration from the menu bar to open the Electronic Configuration dialog. On the
Core Hole tab select 1s from the Shell dropdown list and close
the dialog.
Choose Build | Symmetry | Find Symmetry... from the menu bar to open the Find Symmetry dialog. On the
Options tab check the CoreShellWithHole from the list of properties. On the
Find tab click the Find Symmetry button, then the Impose Symmetry
button. Close the dialog.
Now you should create a core hole on a boron atom and ensure that the symmetry is properly specified.
Repeat these steps for BN_B_hole.xsd.
Select File | Save Project from the menu bar.
4. To set up and run a CASTEP calculation with core holes
You will repeat the previous CASTEP energy calculation determining the core level spectra, but this time taking account of the B and N core holes.
Make BN_N_hole.xsd the active document.
Open the CASTEP Calculation dialog, on the Electronic tab check the Use
core hole checkbox.
Click the Run button, click the Yes button to convert to a primitive cell and close the
CASTEP Calculation dialog.
When the job finishes, the files are transferred back to the client.
Make BN_B_hole.xsd the active document and click the Run button on
the CASTEP Calculation dialog again.
Choose File | Save Project from the menu bar, followed by Window | Close All.
Now you can analyze and visualize the results of the CASTEP calculation.
Make BN CASTEP Energy/BN.xsd the active document and select the N atom.
Choose Modules | CASTEP | Analysis from the menu bar to open the CASTEP Analysis dialog.
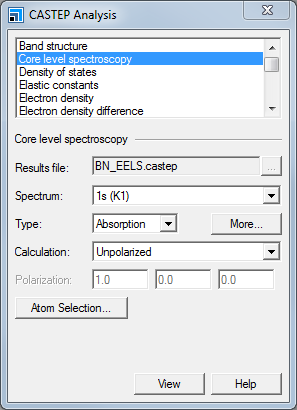
CASTEP Analysis dialog, Core level spectroscopy selection
Select Core level spectroscopy and ensure that BN_EELS.castep is the specified Results file. Select 1s(K1) from the Spectrum dropdown list and choose Absorption for the Type. Click the More... button to open the CASTEP EELS Analysis Options dialog. Set the Instrumental smearing to 0.8 eV and close the dialog.
The 1s[K1] spectrum corresponds to the core level spectrum for a core electron in the 1s orbital. An absorption spectrum simulates the required energy absorbed during the creation of the core hole; an emission spectrum would reflect the energy of an X-ray photon emitted during relaxation of the core hole back to the ground state. The smearing applied by CASTEP is a Gaussian broadening of the calculated results. The value used in this tutorial is selected to correspond to the resolution of the experimental spectra (Jaouen et al., 1995). For a cubic system, such as BN, both polarized and unpolarized incident radiation will produce the same core level spectrum.
Click the View button.
A new core level spectrum for nitrogen in BN is displayed in a chart viewer. Now create the equivalent spectrum for boron.
Select any B atom in BN CASTEP Energy/BN.xsd and, on the CASTEP Analysis dialog, click the View button.
The core level spectrum for boron in BN is displayed. The B and N core level spectra taking into account a 1s core hole can also be created. To ensure that the core hole is used you need to select the correct atom.
Make BN_N_hole CASTEP Energy/BN_N_hole.xsd the active document.
Click the Atom Selection... button on the CASTEP Analysis dialog to open the Atom
Selection dialog. Choose Contains Core Hole from the Select by Property dropdown list
and click the Select button.
You can now view the core level spectrum for the nitrogen atom with the 1s core hole.
On the CASTEP Analysis dialog, click the View button.
Repeat the core hole atom selection and CASTEP analysis for BN_B_hole CASTEP Energy/BN_B_hole.xsd.
6. To compare the results with experimental data
Analysis of the core level spectroscopy properties of BN with and without consideration of a core hole effects has provided you with four spectra, which can be compared with experimentally collected data (Jaouen et al., 1995).
| Spectrum | B | N |
|---|---|---|
| No core hole |
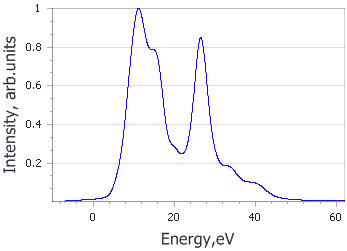
|
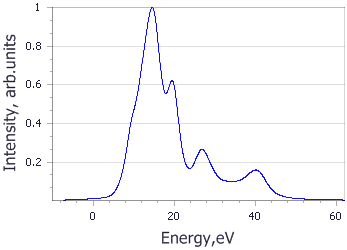
|
| Experiment |
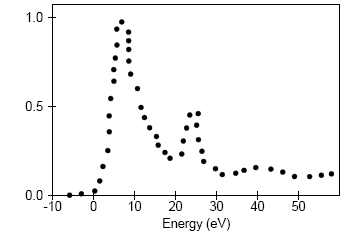
|
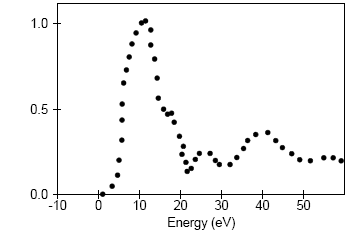
|
| Core hole |
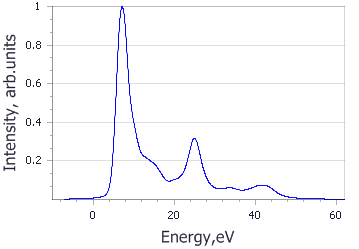
|
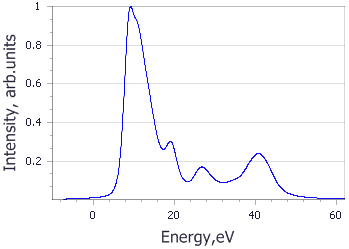
|
This is the end of the tutorial.
References
Gao, S-P.; Pickard, C.J.; Payne, M.C.; Zhu, J.; Yuan, J. "Theory of core-hole effects in 1s core-level spectroscopy of the first-row elements", Phys. Rev. B, 77, 115122 (2008).
Jaouen et al., Microsc. Microanal. Microstruct., 6, 127 (1995)